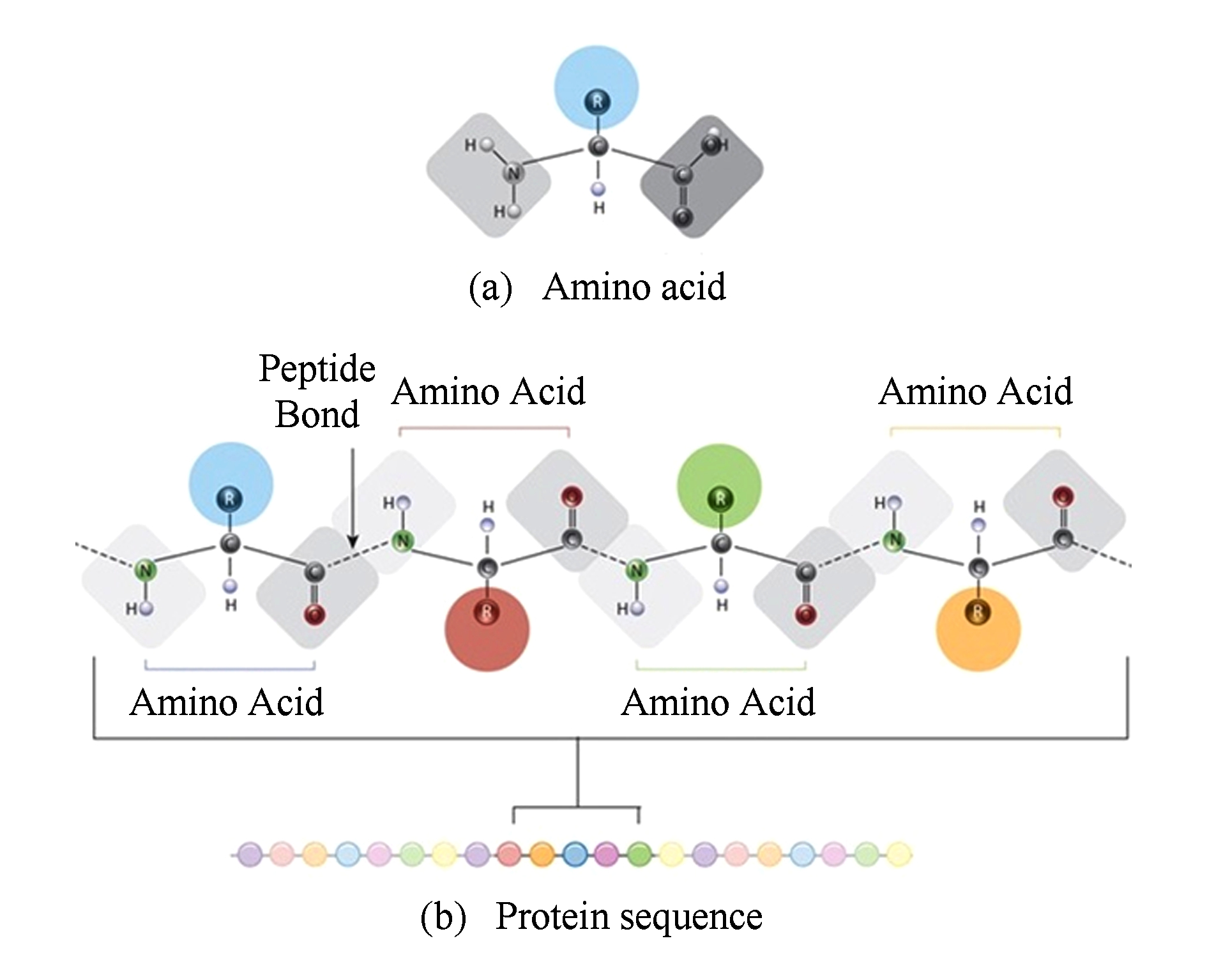
Fig. 1 Illustration of amino acids,peptide bond,and protein sequence
图1 氨基酸、肽键以及蛋白质序列示意图
张海仓 1,2 高玉娟 3 邓明华 3,4,5 郑伟谋 6 卜东波 1
1 (中国科学院计算技术研究所 北京 100190)(中国科学院大学 北京 100049)(北京大学定量生物学中心 北京 100871)(北京大学数学科学学院 北京 100871)(北京大学统计科学中心 北京 100871) (中国科学院理论物理研究所 北京 100190)(zhanghaicang@ict.ac.cn)
摘 要: 蛋白质是由多个氨基酸残基顺序连接而成的长链.在天然状态下,蛋白质并不是无规则的自由状态,而是自发形成特定的空间结构,以执行其特定的生物学功能.驱动蛋白质形成特定空间结构的主要因素是残基间的非共价相互作用,包括疏水作用、静电相互作用、范德华力等.因此,对残基之间远程相互作用的准确预测将有助于对蛋白质空间结构的预测,进而有助于对蛋白质生物学功能的了解.在蛋白质进化过程,有相互作用残基对之间存在一种“共进化”模式,即当一个残基发生变异时,与其有相互作用的残基也要发生相应的变异,以维持相互作用,进而维持整体空间结构以及生物学功能.基于上述生物学观察,研究者开发了多个统计模型和算法以预测残基对之间的相互作用:1)概述残基之间远程相互作用的两大类基本预测算法,包括无监督学习方法和监督学习方法;2)使用蛋白质结构预测CASP比赛结果来客观比较上述各类算法的性能,分析各个算法的特点和优势;3)从生物学观察和统计模型2个角度分析总结了未来的发展趋势.
关键词: 残基远程相互作用预测;蛋白质三级结构预测;图模型;共进化;机器学习
蛋白质是生物体的重要组成成分,行使催化、免疫、细胞信号传导等重要的生物学功能 [1-2] .
蛋白质的基本组成单元是氨基酸,常见的氨基酸有20种.蛋白质是以氨基酸为单元,脱水后由肽键连接而成的长链(氨基酸脱水之后的剩余部分被称为残基).因此从计算的观点看,可以将蛋白质抽象表示成一个字符串序列,其字符集规模为20,其中每一个字符表示一种氨基酸残基,如图1所示:
Fig. 1 Illustration of amino acids,peptide bond,and protein sequence
图1 氨基酸、肽键以及蛋白质序列示意图
在天然环境下,蛋白质呈现的并不是松散的、无规则的形态,而是自发折叠成特定的空间结构,其中每个残基(确切地说是残基中的每个原子)都有其特定的空间坐标.蛋白质的空间结构决定了其生化功能,因此,认识蛋白质的空间结构对了解其功能至关重要 [2] .
目前测定蛋白质结构的主要实验技术包括核磁共振 [3] 、X-ray晶体衍射 [4] 和冷冻电镜 [5] 等.然而上述实验测定技术常常受限于蛋白质大小、蛋白质能否结晶以及结构测定的高成本等因素,使得蛋白质结构的测定速度远远达不到蛋白质序列的测定速度,因而通过计算的方法预测蛋白质结构具有重要的研究意义.另一方面,从序列出发进行蛋白质空间结构预测是可行的 [6] :Anfinsen的经典实验表明在一般情况下,蛋白质折叠是一个自发过程;或者换句话说,蛋白质的结构信息完全蕴含于其序列之中,从而意味着蛋白质结构预测的可行性 [7] .
驱动蛋白质序列形成特定空间结构的主要因素是残基之间的大量非共价相互作用,包括疏水作用、范德华力(van der Waals forces)、离子键以及氢键等.从具有相互作用的残基间序列距离来看,上述相互作用可以分作近程相互作用和远程相互作用2类,其中近程相互作用主导蛋白质形成局部结构,而远程相互作用则引导局部结构的合理摆放,最终形成稳定的蛋白质空间结构 [8] .相对于近程相互作用而言,远程相互作用具有决定整体结构框架的重要作用,从而获得了更多的关注和研究.本文着重讨论残基间远程相互作用预测问题.
蛋白质中残基间是否有相互作用可使用残基之间的欧氏距离作判据,即有相互作用的残基之间距离一般较小.通常采用的标准是:当2个残基的 C β 原子之间欧氏距离小于8Å时,则认为这2个残基具有相互作用,称之为残基间接触(contact),而所有的接触则形象地表示成接触图谱(contact map,见图2).形式上,一个包含 L 个残基的蛋白质接触图谱可以表示成一个 L × L 的矩阵 A * :
(1)
其中, r i 和 r j 分别表示第 i 个残基和第 j 个残基的空间坐标; ![]() 表示该残基对间的欧氏距离; d 表示相互作用残基对之间的距离阈值; sep 表示残基对的序列距离阈值,以此来表示远程相互作用.
表示该残基对间的欧氏距离; d 表示相互作用残基对之间的距离阈值; sep 表示残基对的序列距离阈值,以此来表示远程相互作用.
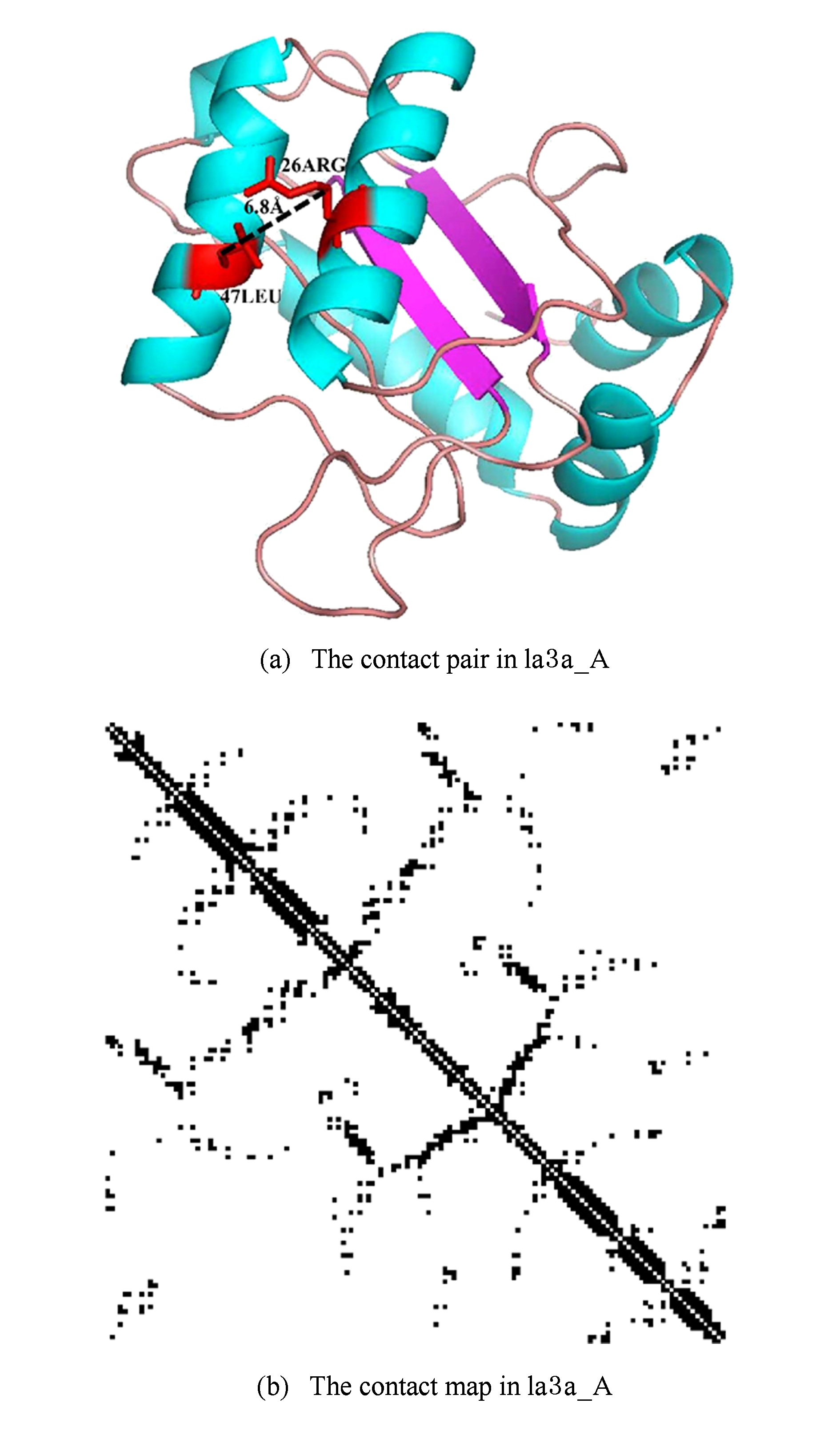
Fig. 2 Illustration of residue-residue contact in a protein 1a3a_A
图2 蛋白质1a3a_A中残基接触示意图
在获得了残基间距离信息之后,采用分子动力学模拟(molecular dynamics simulation, MDS) [9] 等技术可以有效地反推出各个残基的空间位置.因此,残基间相互作用的准确预测成为蛋白质空间结构预测的关键环节之一.
在蛋白质进化过程,有相互作用残基对之间存在一种“共进化”模式,即当一个残基发生变异时,与其有相互作用的残基也要发生相应的变异,以维持相互作用,进而维持整体空间结构以及生物学功能.基于上述生物学观察,研究者提出了多种统计模型和算法以预测残基间相互作用.从统计学的角度讲,由蛋白质序列信息出发预测残基间远程相互作用是一个典型的机器学习问题,即预测组成单元间关联关系的结构学习(structured learning)问题 [10] .
目前的预测方法主要分为2类:无监督学习方法和监督学习方法.简要地说,无监督学习方法仅从序列出发抽取出待测蛋白质的进化历史信息,进而分析2个残基在进化过程中的共变程度,以共变程度的强弱来推断残基间是否存在相互作用.由于这类方法不依赖于已知相互作用的蛋白质集合,因此属于无监督学习方法的范畴.另一类方法是基于已有的结构信息,依据每个残基的序列特征和结构特征,采用神经网络 [11] 、支持向量机 [12-13] 等分类模型预测残基间是否存在相互作用,从而属于有监督学习方法的范畴.
值得强调指出的是在生物信息学这门交叉学科中,重要的是如何将计算模型和生物学现象相结合.具体到残基相互作用这个问题而言:1)残基间相互作用预测大量应用了现有的统计、组合最优化、机器学习等领域的研究成果,是对现有成果的应用和检验;2)残基间相互作用预测问题有其特殊性,主要体现于蛋白质是进化作用的结果.因此,在建模过程中不能简单照搬现有算法和模型,而是需要考虑进化等生物学观察,对现有算法和模型作必要的扩展和改进.
换句话说,每一种统计模型和算法的设计都是基于特定的生物学观察基础之上的,是对生物学观察的数学刻画和描述;另一方面,我们对残基相互作用的生物学观察越深刻,则越有助于我们设计更有效的统计模型.
依据上述观点,本文在介绍每一类算法时,都首先介绍生物学观察,进而介绍如何基于上述生物学观察设计统计模型.
本文综述了目前的蛋白质残基远程相互作用预测算法:1)介绍无监督学习方法(又进一步细分为局部模型和全局模型2类);2)介绍监督学习方法,并以国际蛋白质结构预测竞赛(critical assessment of protein structure prediction, CASP)的结果来分析比较现有方法;3)总结分析未来的发展趋势.
1.1 基本思想
1) 生物学观察.从进化的角度来看,同源蛋白质是指由同一个祖先蛋白质进化而来的后代蛋白质,其结构和功能有一定的保守性,这种保守性由残基的远程相互作用来维持.在进化过程中,当有相互作用的残基对中其中一个残基发生突变时,另一个常常也发生相应的突变,以维持相互作用,否则不利于蛋白质整体结构的稳定.这种现象被称之为“有相互作用残基对的共进化”,如图3所示.
2) 数学模型设计.通常采用多序列联配的数学形式来刻画同源蛋白质之间的同源关系,其中每一列表示由祖先蛋白质中的某个残基进化生成的残基(如图4所示).考虑长度为 L 的多序列联配,通常用离散随机变量 X i (1≤ i ≤ L )表示第 i 个残基(亦称为位点)的氨基酸种类,可取21个离散值(包括20种氨基酸和一个联配空位),则多序列联配中的每条序列可看成这些变量的一个观测样本.

Fig. 3 The phylogenetic tree and MSA of PF00111
图3 序列家族PF00111的进化树和多序列联配
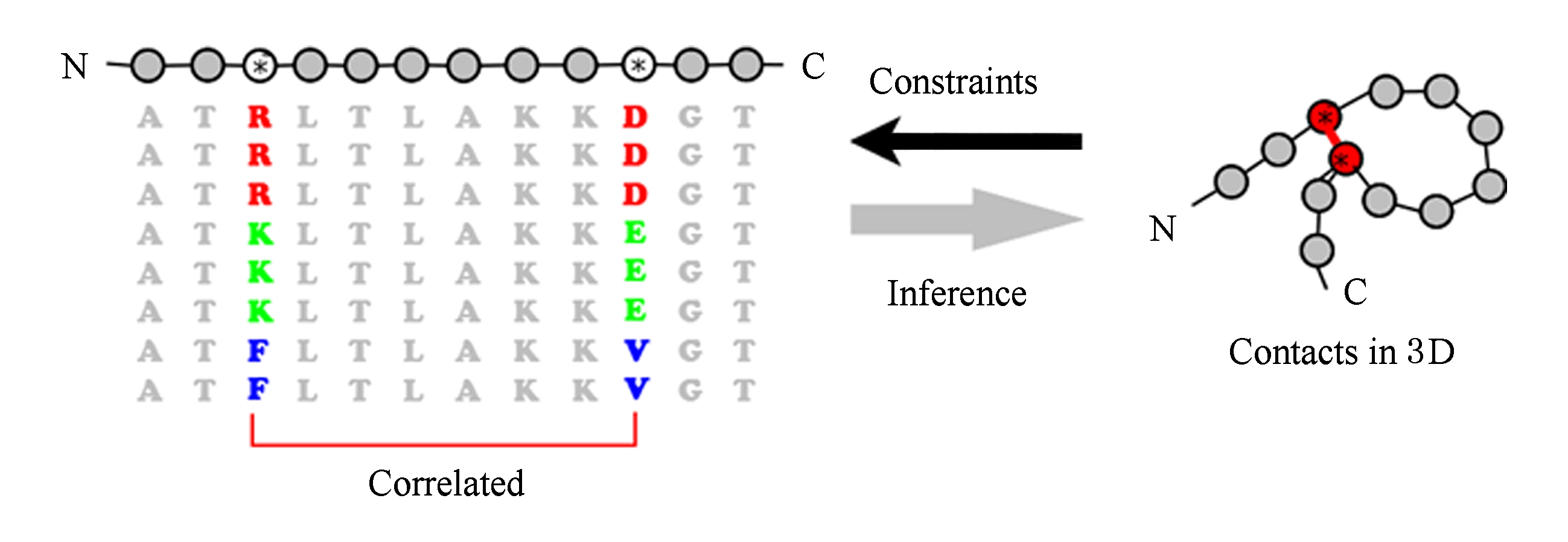
Fig. 4 Illustration of the principal for contact prediction using unsupervised methods
图4 无监督学习方法预测残基远程相互作用原理示意图 [14]
基于残基共进化现象,无监督学习方法的基本思想是首先检索出与待预测蛋白质序列同源的所有序列,计算出多序列联配(multiple sequence alignment, MSA),以此来表示待测蛋白质的进化历史信息;进而分析2个残基在进化过程中的共进化(或者共变异)程度,以共变程度的强弱来推断残基间是否存在相互作用.从统计的角度来讲,即是通过多序列联配中列向量之间的相关性,即2个随机变量 X i , X j 之间的相关性,从而推断残基之间的相互作用.
无监督学习方法可以分为两大类,即局部模型和全局模型 [6] .其中,局部模型假设一个残基对内部的相关性与其他残基对是独立的,从而每对残基单独计算其相关性;而全局模型则考虑了残基对之间的关联关系,对所有的残基建立统一的全局模型.我们在本节介绍局部模型,在第2节介绍全局模型.
1.2 典型方法
局部模型在计算某对残基之间的相关性时,不考虑其他残基对的影响,直接分别计算各残基对之间的相关性;各种局部模型的差异主要体现在变量相关性的衡量方法不同.下面我们介绍3种典型的局部模型,并分析其优缺点.
1.2.1 典型方法1:共变相关系数
Gobel等人 [15] 提出第1个预测残基相互作用的方法——相关系数法,该方法通过计算蛋白质多序列联配中2列间的Pearson相关系数,通过对不同位点突变的相似程度来定量刻画共进化程度,进而推断具有相似变异特征的残基对.其基本思想是:首先基于多序列联配,对每一个位点 i 由氨基酸替换打分计算得到替换矩阵,记为 s i ,其中元素 ![]() 是第 k , l 条序列在第 i 位点处的残基替换得分;然后对任意2个位点,使用上述残基替换矩阵计算共变相关系数:
是第 k , l 条序列在第 i 位点处的残基替换得分;然后对任意2个位点,使用上述残基替换矩阵计算共变相关系数:
(2)
其中, M 是多序列联配的序列条数,〈·〉为矩阵元素均值, σ i 为矩阵 s i 所有元素的标准差,如图5所示.
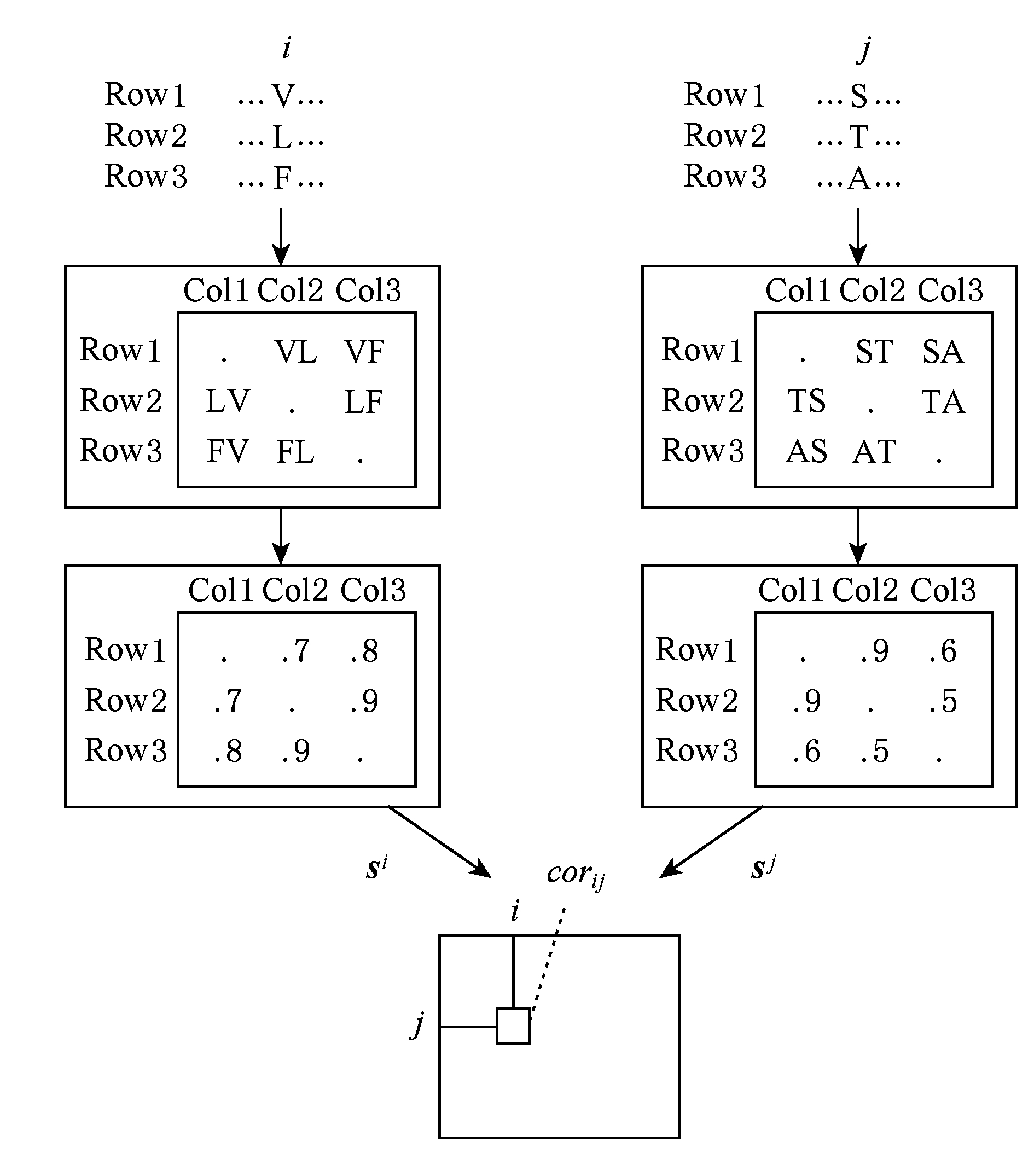
Fig. 5 Illustration of co-mutation extraction for residue pair (i,j) [15]
图5 位点对(i,j)共变信息计算示意图 [15]
实验表明:将相关系数作为共变度量,在一定阈值下推断残基间相互作用,准确率比随机预测有显著提高,从而表明由残基共变性推断其相互作用的可行性;然而Pearson相关系数只能度量随机变量间的线性相关关系,从而使得该方法存在一定的局限性.
1.2.2 典型方法2:互信息
Martin等人 [16] 用互信息(mutual information, MI)识别共进化残基对.这种方法源于信息论,具体来说,对于某个多序列联配中的位点 i 和 j ,其互信息定义为
(3)
其中, f ij ( a , b )为位点 i 出现残基 a 且位点 j 出现残基 b 的频率, f i ( a )表示位点 i 出现残基 a 的频率.与Pearson相关系数相比而言,互信息可以度量变量之间的非线性关系,其值越大表示残基对间的共进化程度越大,互信息为0则表示2位点独立进化,或存在保守位点.
Martin等人的实验结果表明互信息较高的位点对倾向于具有相互作用,但是其效果受限于序列条数和进化偏差造成的背景噪音.因而欲提高预测准确率,需降低背景噪声的影响,减少对序列条数的依赖,从而为后续研究指明了改进方向.
1.2.3 典型方法3:OMES
Kass等人 [17] 提出另外一种共变性的度量方法OMES(observed minus expected squared),这种方法基于统计学中的卡方检验,通过比较残基对在2个位点上实际出现次数与期望出现次数之间的差异来定量刻画残基对的共进化程度.具体地,其定义为
(4)
其中,O ij ( a , b )和E ij ( a , b )分别表示残基对( a , b )出现频数的观测值和期望值, M 是序列条数.O ij ( a , b )可以直接从多序列联配中统计得到;E ij ( a , b )是在假设残基对间不存在相关性的前提下计算得到的,即E ij ( a , b )= Mf i ( a ) f j ( b ),其中 f i ( a ), f j ( b )分别表示相应位点某氨基酸的出现频率.OMES的值越大表示2个位点之间的共进化程度越高;对于2个完全独立进化的位点,OMES的值为0.
我们在GREMLIN数据集 [18] 上测试OMES方法,并与MI进行了比较;实验结果表明:MI与OMES的预测性能相当,详细实验结果分析参见第4节.
1.3 局部模型的实验结果及分析
局部模型的优点是简单、计算速度快;但是也有较大的不足,主要表现为:1)各残基对之间并不是独立的,而是存在关联传递的现象,局部模型并没有考虑这种关联传递现象;2)未考虑序列空间采样偏差及样本数不足的影响;3)相关性计算存在大量由进化偏差产生的背景噪声.
虽然局部模型普遍存在预测准确率偏低的缺陷,然而在一定程度上提取了进化信息,是由序列信息推断结构约束的早期尝试,对后续研究具有重要的启发和借鉴意义.
2.1 基本思想
1) 生物学观察.局部模型单独计算各个残基对的相关性,其暗含的假设是某个残基对的相互作用是独立于其他残基对的.然而一个残基可能与多个残基有作用,从而导致关联传递这一普遍存在的现象.如图6所示,如果残基 A 和残基 D 共变,残基 D 和残基 C 共变,那么从序列信息来看,残基 A 和残基 D 也表现出共变性,然而残基 A 和残基 D 之间的共变性源于传递效应,并非源自残基 A 和残基 D 的相互作用.这种通过残基共进化的传递效应导致的相关性称为间接关联.
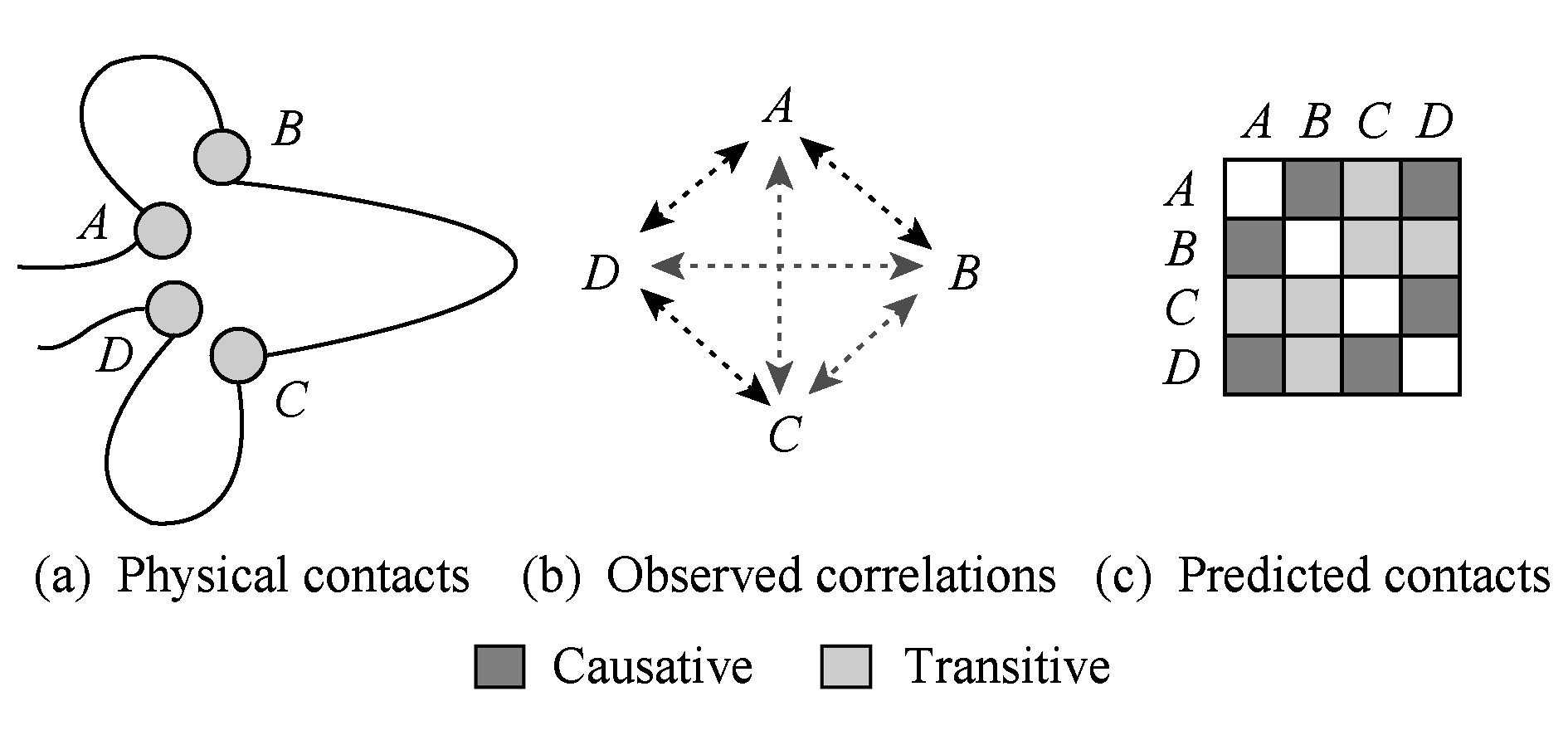
Fig. 6 Illustration of direct and indirect couplings
图6 直接关联和间接关联示意图 [25]
2) 数学模型设计.局部模型假设任意2个残基的共进化和其他残基是相互独立的,决定了它只能探测相关性,会受到间接关联噪声的显著影响,从而不能准确提取出真实共进化(接触)的残基对.鉴于此,全局模型对所有位点建立全概率模型,同时考虑所有残基对之间的关联关系,试图去除间接关联的影响,从而避免局部模型的缺陷.
2.2 典型方法
迄今为止已经发展了多种全局模型,比如Markov随机场模型(Markov random fields, MRF) [19-21] 、贝叶斯网络模型(Bayesian network) [22] 、高斯图模型(Gaussian graphical model) [23-24] 和网络反卷积(network deconvolution)等 [25] .这些方法的不同之处主要体现在如何对多序列联配建模,其中贝叶斯网络模型采用有向图模型进行建模;Markov随机场模型和高斯图模型采用无向图模型进行建模;高斯图模型可以看成Markov随机场模型的特殊形式.
2.2.1 典型方法1:贝叶斯网络模型
Burger等人提出使用贝叶斯网络模型预测残基间相互作用 [22] .在这种方法中,使用贝叶斯网络把残基间的共进化关联关系表示成依赖关系:当位点 i 和位点 j 存在相互作用时,则第 i 个位置出现残基 X i 的概率依赖于第 j 个位置出现残基 X j 的概率.这种依赖关系形象地表示成贝叶斯网络中的一条有向边,如图7所示.
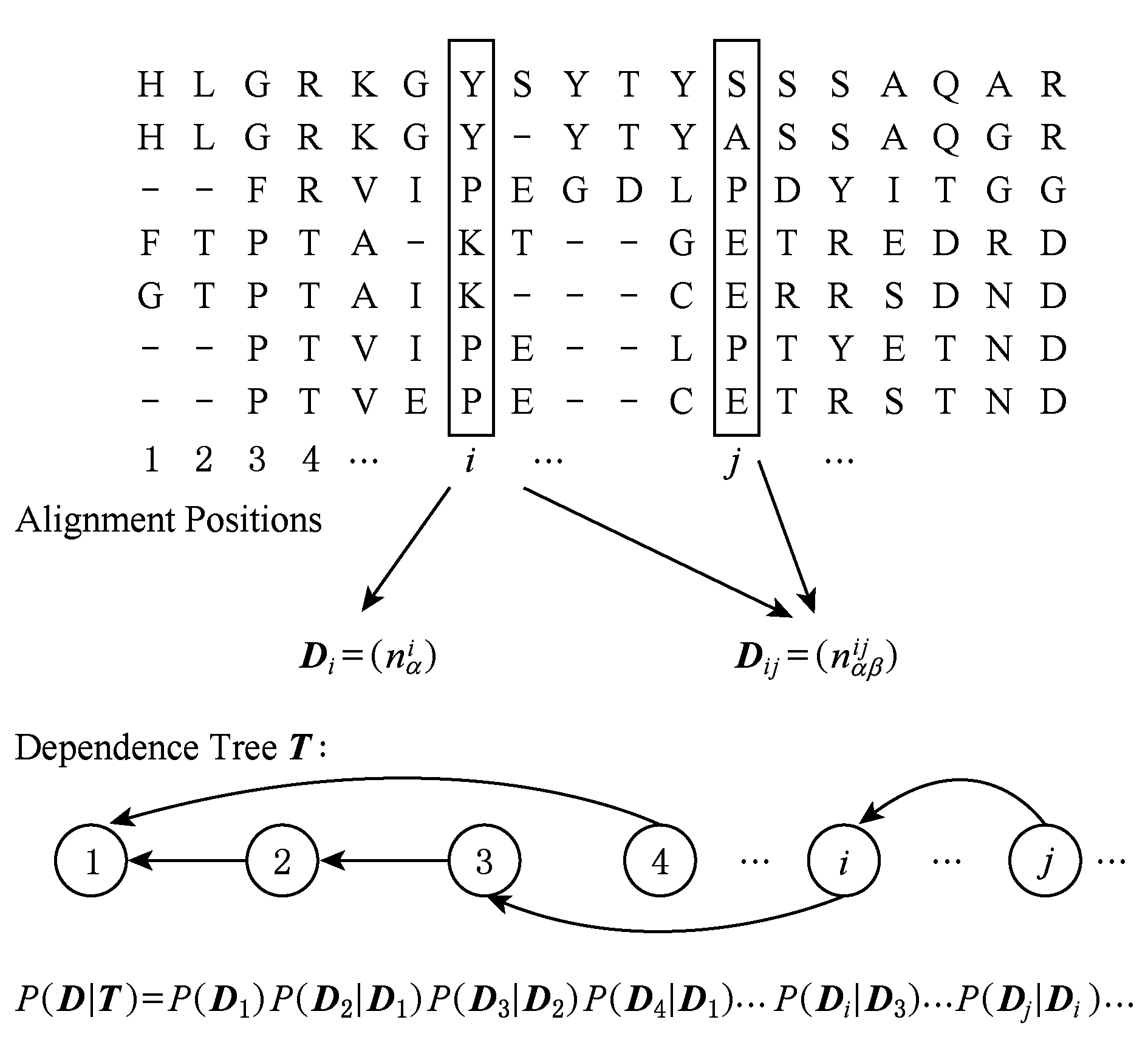
Fig. 7 Bayesian network model of a given MSA
图7 贝叶斯网络模型示意图
在已知位点之间依赖关系的情况下,可以计算观察到某个多序列联配的条件概率;反过来,在给定多序列联配的情况下,结合依赖关系的先验分布,可以推断位点 i 和位点 j 之间存在依赖关系(共进化)的后验概率,最后认为后验概率高的残基对具有相互作用.
本节首先描述给定依赖关系的情况下观察到某个多序列联配的条件概率计算过程,然后介绍依赖关系的后验概率的计算过程.
1) 已知残基间依赖关系计算多序列联配的条件概率
假设残基间所有的依赖关系形成有向图 T =( π , V ),其中 π 表示所有有向边的集合, V ={1,2,…, L }表示残基位点集合.为简化计算,进一步假设依赖关系图 T 是树状图,即在 T 中存在唯一根节点 r ,除根节点 r 外,其余节点 i 都存在唯一父节点,记其父节点为 π ( i ).
给定残基间依赖关系 T ,则观察到多序列联配 D 的条件概率为
![]() ,
,
(5)
其中, S ij = P ( D i , D j )  ( P ( D i ) P ( D j ));
( P ( D i ) P ( D j )); ![]() ,表示MSA第 i 列中各氨基酸出现频率向量.假设第 i 列中氨基酸 α 出现的概率为
,表示MSA第 i 列中各氨基酸出现频率向量.假设第 i 列中氨基酸 α 出现的概率为 ![]() ,且 w i 服从Dirichlet分布,则:
,且 w i 服从Dirichlet分布,则:
P ( D i )=∫ P ( D i | w ) P ( w )d w =
![]() ,
,
(6)
同理可得:
![]() ,
,
(7)
其中参数 λ , λ ′是伪计数.
2) 残基间依赖关系后验概率的计算
假设依赖关系图 T 的先验分布为 ![]() (1- μ e ),其中, μ e 表示依赖关系 e 出现的概率,通常由多序列联配中相应2列的互信息等估计.记
(1- μ e ),其中, μ e 表示依赖关系 e 出现的概率,通常由多序列联配中相应2列的互信息等估计.记
(8)
则式(8)可重写为
![]() .
.
(9)
由式(9)可推出MSA的概率模型为
![]() ,
,
(10)
其中, B jπ ( j ) = S jπ ( j ) W jπ ( j ) .
注意到式(10)中的 ![]() B jπ ( j ) 是对所有树求和,若简单采用枚举策略则需要指数多的时间.一种简化的计算方法是依据Kirchhoff定理 [26] ,有
B jπ ( j ) 是对所有树求和,若简单采用枚举策略则需要指数多的时间.一种简化的计算方法是依据Kirchhoff定理 [26] ,有 ![]() B jπ ( j ) =det( Q ( La )),其中 La 表示矩阵 B 的Laplacian矩阵
B jπ ( j ) =det( Q ( La )),其中 La 表示矩阵 B 的Laplacian矩阵 ![]() - B ij , Q ( La )表示将矩阵 La 去掉第1行和第1列后的矩阵.
- B ij , Q ( La )表示将矩阵 La 去掉第1行和第1列后的矩阵.
使用贝叶斯公式计算 T 的后验分布为: P ( T | D )= P ( D | T ) P ( T ) P ( D ),进而对于特定的残基对( k , l )之间有边 E kl 的后验概率通过对包含 E kl 的所有树的后验概率求和得到,如图8所示:
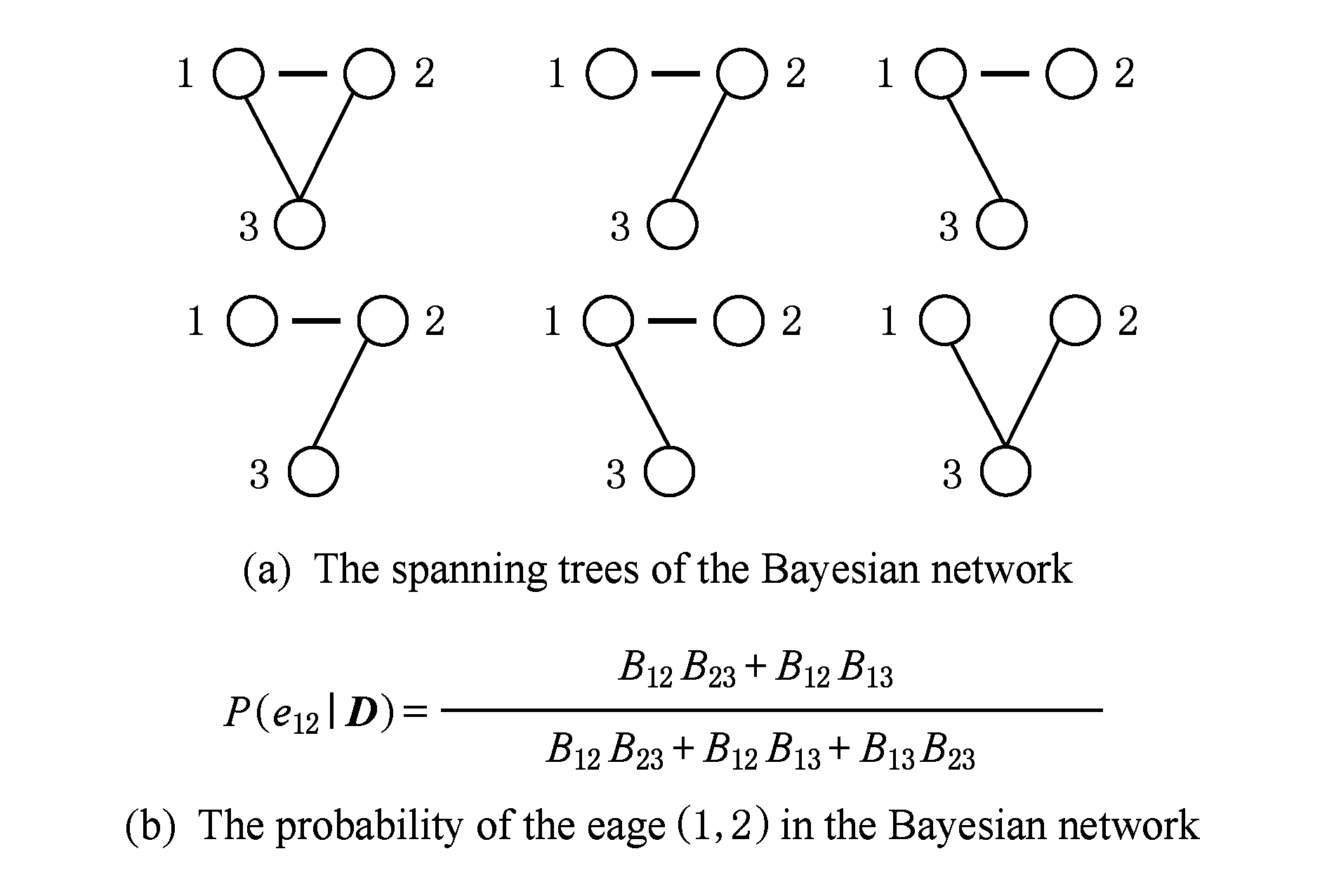
Fig. 8 Illustration of the calculation of posterior probability
图8 后验概率计算示意图 [22]
P ( E kl | D )=
![]() ,
,
(11)
其中, ![]() 同样可由Kirchhoff定理计算.
同样可由Kirchhoff定理计算.
实验结果表明:以后验概率 P ( E ij | D )作为残基对( i , j )的相关度量能够去掉间接关联,和局部模型相比,显著提高了预测准确率.
2.2.2 典型方法2:Markov随机场
Markov随机场是一种无向图模型,其形式可由最大熵原理推导得到,所以也被称为最大熵模型.Markov随机场的优势在于可以直接刻画残基间的远程相互作用 [19-21,27] .
设多序列联配的长度为 L ,变量 X i 表示第 i 个位置出现的氨基酸,则多序列联配中的一条序列的生成概率为
P ( X 1 , X 2 ,…, X L )=
![]() ,
,
(12)
其中:
为配分函数, e ij ( X i , X j )表示位置 i 处氨基酸 X i 和位置 j 处氨基酸 X j 的耦合强度, h i ( X i )为位置 i 处观察到残基 X i 的可能性,均为待确定的参数.最终的推断规则为:耦合强度强的残基对被预测为具有相互作用.
假设给定包含 M 条序列的多序列联配,上述待定参数 e ij ( X i , X j )与 h i ( X i )可以通过极大似然策略进行估计.然而上述极大似然策略涉及到配分函数 Z 的计算,其计算是NP难问题,所以发展出多种近似求解方法,包括置信传播算法、平均场近似算法和伪似然最大化算法,简要介绍如下:
1) 置信传播算法bpDCA
Weigt等人 [20] 用Markov随机场模型研究蛋白质-蛋白质相互作用,并用置信传播算法(bpDCA)近似求解模型参数,后来这种方法也被直接借用于残基间相互作用的推测.
置信传播算法的基本思想是通过局部信息的多次传播以逼近全局信息,以此计算配分函数或边际概率.确切地说,在最大化似然函数过程中,梯度函数的计算涉及边际概率,而置信传播算法的核心是解决梯度计算问题.在给定初始参数的情况下,bpDCA迭代执行2个步骤直至满足收敛条件:
① 用置信传播算法估计边缘分布 P i ( X i )和 P ij ( X i , X j )
首先对于每个位置 i ,迭代求解信息传递 P i → j ( X i ):
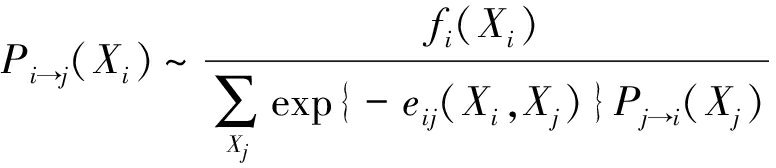 ,
,
(13)
此处 f i ( A )为经验频率.
然后可获得边际分布 P i ( X i )的估计:
P i ( X i )~exp{ h i ( X i )}
![]() .
.
(14)
用类似的方法可得 P ij ( X i , X j )的估计.
② 用梯度下降策略更新参数估计
首先,似然函数的梯度可估计为
Δ e ij ( X i , X j )= f ij ( X i , X j )- P ij ( X i , X j )-
![]() ,
,
Δ h i ( X i )= f i ( X i )- P i ( X i ),
(15)
其中, f i ( A )和 f ij ( A , B )为经验频率.然后更新参数估计为
![]() ,
,
![]() ,
,
这里 ρ 为迭代步长.
bpDCA主要有2个缺陷:①速度慢.该算法迭代1次的计算复杂度为 O (21 2 L 4 ),即使对长度为60的短蛋白,bpDCA在4核CPU上也需大约运行4 d.②收敛性差.该算法解的渐进性质不能得到有效控制,理论上不能保证其收敛性.
2) 平均场近似算法mfDCA
Morcos等人 [19] 提出使用平均场近似策略(mfDCA)来近似求解Markov随机场的参数.平均场近似的基本思想是由简单可分解分布近似复杂分布,因此其核心在于2个问题:①如何确定简单分布的形式;②如何衡量简单分布和复杂分布之间的差异,并找到最接近原始复杂分布的简单分布.
一种常用的近似方法是:假设简单分布是完全可分解的分布,即 ![]() P(X i ),并用KL距离度量可分解分布和原分布之间的差异 [10] .mfDCA则采用了Small-coupling expansion的方法 [28-29] ,对Markov随机场分布的Gibbs能量进行泰勒一阶近似,最后通过对经验协方差矩阵求逆推断出耦合参数,即
P(X i ),并用KL距离度量可分解分布和原分布之间的差异 [10] .mfDCA则采用了Small-coupling expansion的方法 [28-29] ,对Markov随机场分布的Gibbs能量进行泰勒一阶近似,最后通过对经验协方差矩阵求逆推断出耦合参数,即
(16)
其中 C 可由经验协方差矩阵计算,即 C ij ( A , B )= f ij ( A , B )- f i ( A ) f j ( B ).此处 f i ( A )和 f ij ( A , B )为经验频率.
mfDCA的优势是速度快,通过求逆计算耦合参数的时间复杂度是 O ( L 3 ),比bpDCA速度提高上千倍,从而使大量蛋白质家族的计算成为可能.
3) 极大伪似然算法plmDCA
Lövkvist等人 [21] 提出伪似然最大化方法估计MRF的参数,其基本思想是用伪似然函数近似似然函数.由于计算伪似然函数梯度的时间复杂度是多项式的,所以可以有效地估计参数.
伪似然函数定义如下:
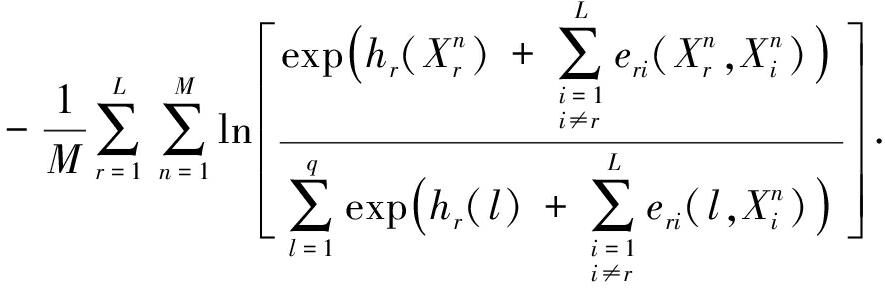
(17)
上述模型参数数目过多,对长为 L 的序列来说,模型参数规模为(20×20)× L ( L -1) 2+20 L .当 L =100时,模型将有近200万的参数.为避免过拟合问题,Ekeberg等人在伪似然函数中引入了正则项 R ( h , e ),即通过解决以下优化问题求解参数:
{ h PLM , e PLM }=arg min{ l pseudo ( h , e )+ R ( h , e )},
(18)
![]() .
.
其中, λ h 和 λ e 分别表示单体项 h 和双体项 e 的正则化参数.
该方法避免了极大似然求解复杂配分函数的问题且当样本量足够大时,极大伪似然估计是极大似然估计的一致估计 [30] ,从而能够保证获得准确的参数估计,并且和mfDCA相比显著提高了准确率.
Kamisetty等人 [18] 在极大伪似然的基础上,进一步将结构先验信息引入正则项,开发软件GREMLIN.实验结果表明,由于引入结构先验信息,GREMLIN方法的性能优于plmDCA.
2.2.3 典型方法3:高斯图模型
高斯图模型假设多序列联配中每一条蛋白质序列都服从高斯分布 N ( μ , Σ ),其中高斯分布的协方差矩阵的逆称为精细矩阵(recision matrix),记作 Θ = Σ -1 .精细矩阵表征了变量之间的直接关联信息 [31] ,因而可以通过精细矩阵来预测残基间的相互作用.在统计学中,通过分析精细矩阵来推断直接关联的策略也称为偏相关分析.
为防止过拟合,通常采用引入正则项的策略来控制模型的复杂度.典型的方法包括PSICOV所使用的图Lasso以及CoinDCA所采用的成组Lasso,简要介绍如下:
1) PSICOV使用的图Lasso策略
Jones等人 [24] 利用图Lasso策略推断精细矩阵,开发了软件PSICOV.该方法的核心思想是优化含有正则项的似然函数:
(19)
其中, COV 为经验协方差阵.前2项为高斯分布的对数似然,第3项为正则项.正则项的引入有2个作用:①控制模型复杂度,防止过拟合,以避免模型参数过多导致参数推断的困难;②保证精细矩阵的稀疏性,以此刻画接触图谱的稀疏性.
Jones等人 [24] 在150个目标蛋白进行测试,结果显示PSICOV一致优于局部模型和贝叶斯网络模型.
2) CoinDCA采用的成组Lasso策略
Ma等人 [23] 对高斯图模型做了扩展,以融合多个相关家族的进化信息,并开发了软件CoinDCA.
CoinDCA的基本思想是:假设对于目标蛋白质序列,与其具有相同折叠类型的共有 K 个家族(可通过同源搜索获得),由于这 K 个蛋白家族属于同一折叠类型,所以可认为它们有类似的蛋白质接触图谱;相应地,当用 K 个高斯分布分别对 K 个蛋白家族建模时,它们有类似的精细矩阵;成组Lasso的目的是约束这 K 个高斯分布具有类似的精细矩阵.
具体地,通过优化下式求解这些精细矩阵.
(20)
其中,前3项和PSICOV类似,这里是 K 个蛋白家族带正则项的联合似然函数; ![]() 2 =
2 = ![]() 通过将各个家族相关列对应的变量放在同一组,使得 K 个精细矩阵具有类似结构.
通过将各个家族相关列对应的变量放在同一组,使得 K 个精细矩阵具有类似结构.
CoinDCA充分利用了相近家族的进化信息且融合了监督学习(随机森林)的方法.在PSICOV,CASP10和CASP11数据集上测试的实验结果表明,这种方法对同源序列少的蛋白预测准确率有显著提高,减少了对同源序列数目的依赖;而单一地整合多家族信息或者采用随机森林的方法并不能对预测性能有所改进.
2.2.4 典型方法4:网络反卷积
在残基相互作用中消除间接效应,本质上是网络推断领域直接作用和间接作用的区分问题 [32] .Feizi等人 [25] 提出网络反卷积(network deconvolution, ND)策略推断直接相互作用网络.
网络反卷积的基本思想是:假设观测到的相关矩阵 G obs 是直接相关矩阵 G dir 和间接相关矩阵的叠加,而间接相关可视为直接相关通过多步传递得到的(如图9所示),即:
![]() ,
,
注意到当矩阵 G dir 特征值绝对值小于1时,等式右边收敛,上式有闭合形式:
(21)
所以可用简单代数策略由观测相关矩阵 G obs 通过网络反卷积可得到直接相关矩阵 G dir .为保证式(18)的收敛性, G dir 的特征值需满足| λ dir |<1.相应地, G obs 的特征值需满足 λ obs >-0.5.所以需对原始观测矩阵 G obs 进行线性缩放 ![]() = αG obs ,对
= αG obs ,对 ![]() 进行反卷积得到直接相关矩阵 G dir .
进行反卷积得到直接相关矩阵 G dir .
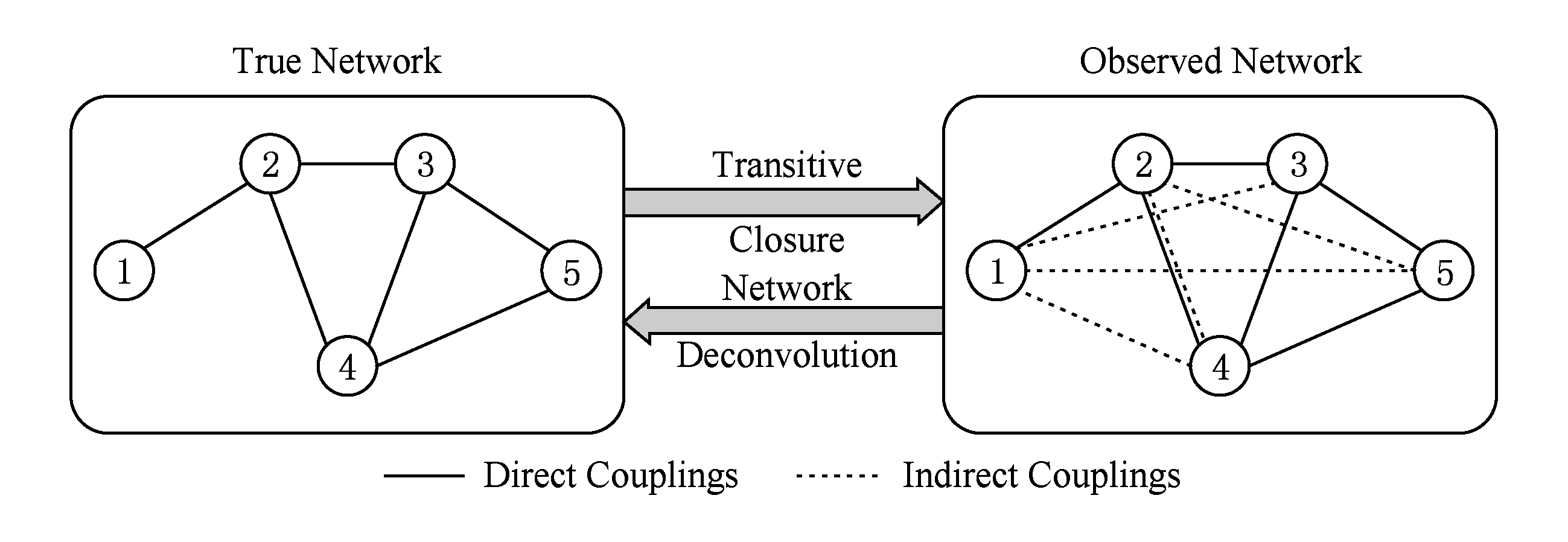
Fig. 9 Illustration of network deconvolution
图9 网络反卷积意图 [25]
网络反卷积方法广泛应用于社交网络、基因调控网络等领域的推断中.Wright等人 [32] 在残基相互作用推断中的结果表明,网络反卷积策略可有效过滤掉互信息的间接关联噪声,而对全局模型mfDCA输出的直接信息(已去除间接关联)进行反卷积并没有显著改进;而对互信息矩阵反卷积的预测效果不如mfDCA.上述结果说明网络反卷积的策略具有普适性,但对于特定的残基相互作用预测问题则仍然需要进行相应的改进.
Sun等人 [33] 提出了平衡网络反卷积方法,该方法不需要像原始网络反卷积方法那样对 G obs 进行线性缩放,其假设
G obs = G dir + G indir =
![]() ,
,
(22)
则:
(23)
可以导出对任意的 λ obs ∈(-∞,+∞),都有| λ dir|<1,所以该方法不需要对 G obs 进行线性缩放.结果表明,平衡网络反卷积方法的预测性能优于原始网络反卷积方法;如何进一步提升预测性能,仍然需要后续有针对性的研究.
2.3 全局模型的实验结果及分析
我们在GREMLIN数据集上详细测试了无监督学习方法的性能,如表1、表2所示,结果显示全局模型优于局部模型,且在全局模型中plmDCA预测性能最优.具体地,我们从实验结果分析中获得3个结论:
1) 由于有效地去除了间接效应的影响,全局模型比局部模型革命性地提高了预测准确率.
2) 全局模型之间的预测性能差距相对较小,且不同方法的预测结果有一定程度的不同,将不同方法得到的残基相互作用信息有效地整合,并用于蛋白质三级结构预测,具有重要意义和广泛的应用前景.
Table 1 Denoising Performance of LRS for Three LocalMethods on GREMLIN Benchmark
表1 在GREMLIN测试集上LRS对局部模型去噪效果
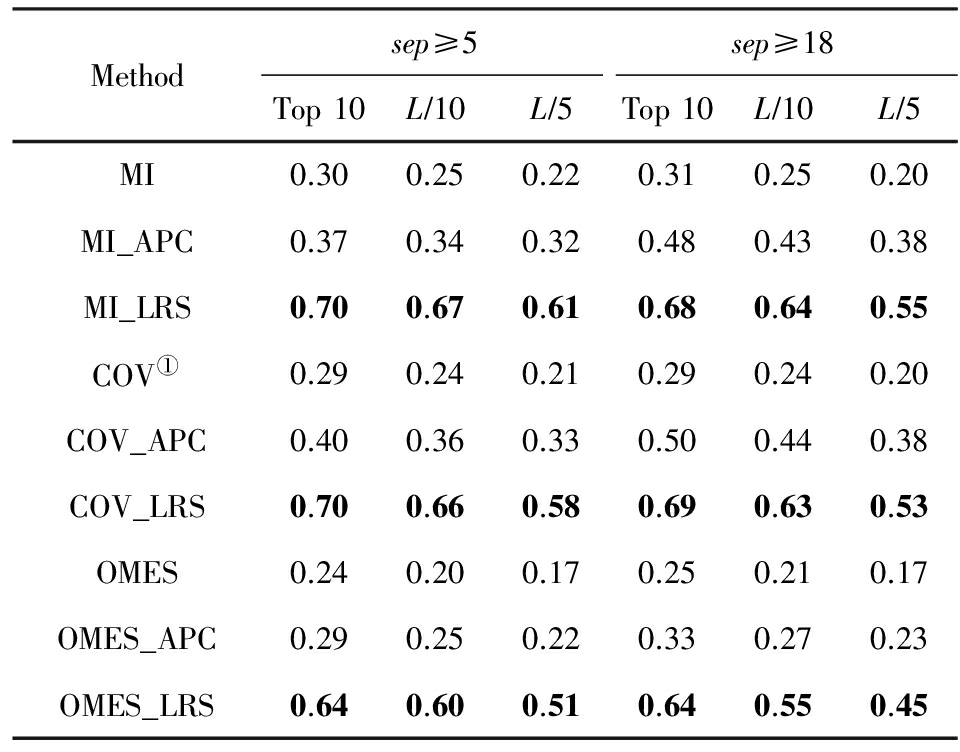
① ![]()
Table 2 Denoising Performance of LRS for Three Global Methods on GREMLIN Benchmark
表2 在 GREMLIN 测试集上 LRS 对全局模型去噪效果
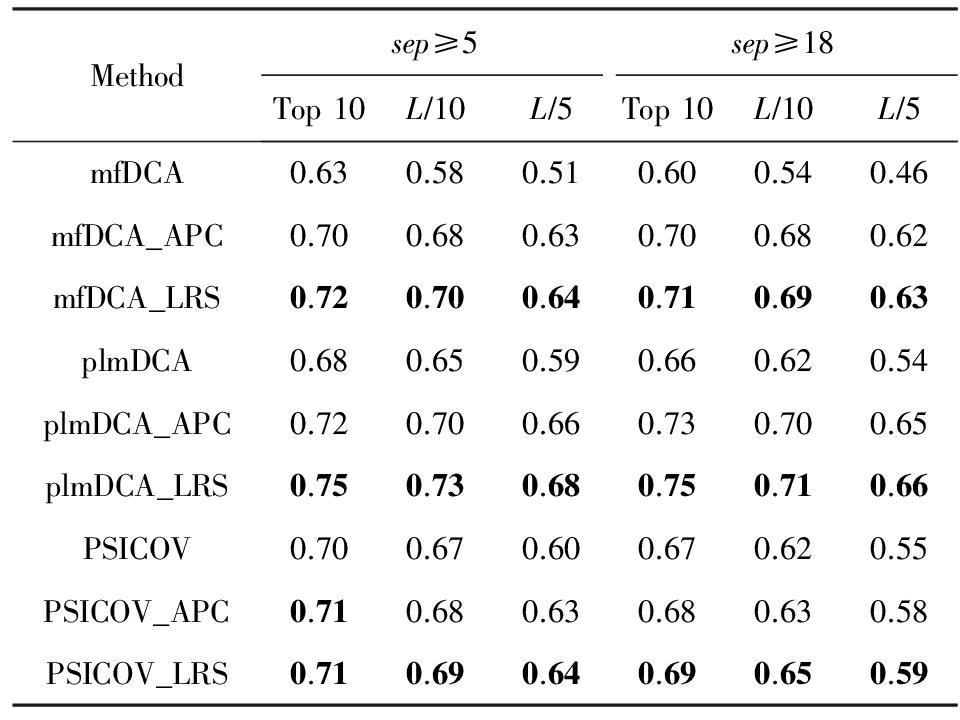
3) 全局模型普遍存在参数多的问题,要得到精确的参数估计需要大量样本信息,预测效果强烈依赖于同源序列的数目;且复杂的参数估计导致全局模型运行速度较慢,需要开发更加有效的参数估计方法.
除了统计模型之外,影响远程相互作用预测性能的因素还包括样本的不独立性以及背景噪声的影响等,因此需要采取预处理和后处理步骤以消除这些因素的影响.通常采用的预处理和后处理步骤简要陈述如下.
3.1 克服样本不独立性的预处理过程
多序列联配中的样本是与待测蛋白同源的序列,这些序列来源于同一祖先蛋白质,从而造成了观测样本之间的不独立性,影响模型预测的准确性.为提高准确性,一般对多序列联配进行2方面预处理:
1) 去冗余.比如去掉与目标蛋白质高度相似(通常采用序列等同度大于90%)的序列.
2) 加权重.对于每条序列,都依据在多序列联配中与其相似的序列条数赋予权重,其基本思想是:如果一条蛋白质序列具有较多的相似序列,则权重较低;反之则设置较高权重.具体地,对第 k 条序列来说,首先统计与其序列等同度高于80%的序列数目:
m k =|{ n ∈{1,2,…, M }| seqid ( X k , X n )>80%}|,
(24)
进而在统计残基和残基对频率向量时,将每条序列的权重设置为1 m k .
3.2 去背景噪声的后处理过程
同源序列中通常包含由进化造成的较强的背景噪声.具体来说,如果一个位点突变发生在进化早期,其后代都将延续这个突变,从而导致过高地估计了此位点和其他位点之间的共变性.
通常采用后处理的方法消除这些背景噪声对相互作用预测的影响,常用的策略简要介绍如下:
1) 均值乘积校正方法(average product correction, APC)
Dunn等人 [34] 基于信息论提出对互信息矩阵进行归一化去噪的方法APC.其基本思想是假设背景噪声具有如下的均值乘积的形式:
![]() .
.
(25)
其中 MI i .表示位点 i 与其他位点互信息的平均值; MI ..表示所有位点对互信息的平均值.
经APC去噪后的互信息 MIp 为
MIp ij = MI ij - APC ij .
(26)
实验结果表明采用APC技术去除背景噪声后,能够有效提高基于互信息对残基相互作用预测的精度.目前,该策略被推广应用到其他相关矩阵,已成为全局统计模型标准的后处理步骤.
2) 谱去除方法(spectrum cleaning, SC)
Halabi等人 [35] 假设背景噪声存在于相关性矩阵的第一主成分里,其依据是第一主成分表示相关性的整体一致性,能够刻画由进化偏差引起的背景噪声.具体地,对于采用局部模型或者全局模型计算出的相关性矩阵 M 进行奇异值分解 ![]() ,其中 r 为矩阵 M 的秩;谱去除方法认为背景噪声可以表示为
,其中 r 为矩阵 M 的秩;谱去除方法认为背景噪声可以表示为 ![]() 1,则去噪后的相关矩阵为 Mp =
1,则去噪后的相关矩阵为 Mp = ![]() .
.
3) 低秩稀疏矩阵分解方法(low rank and sparse matrix decomposition, LRS)
谱去除方法假设背景噪声来源于相关性矩阵的第一主成分,其秩为1;然而当多序列联配中的序列是来源于多个家族,则相关性矩阵的其他主成分也会含有背景噪声 [35] .基于上述认识和观察,我们团队假设背景噪声是低秩的,同时真实的相互作用是稀疏的.大量统计数据支持上述假设的合理性,即真实相互作用仅占所有可能相互作用的3%~5% [11,24] .基于上述生物学观察,我们设计了统计模型将背景噪声和真实的相互作用信号区分开来 [36] .
具体地,对于给定的残基相关性度量矩阵 M ,我们认为它是低秩噪声矩阵和稀疏信号矩阵的叠加,进而用低秩稀疏矩阵分解技术还原真实的相互作用信号矩阵,即求解如下的优化问题:
(27)
其中 ![]() σ i ( L )为谱范数,以衡量背景噪声矩阵的秩;
σ i ( L )为谱范数,以衡量背景噪声矩阵的秩; ![]() | S ij |度量相互作用矩阵的稀疏性; λ 控制稀疏和低秩的相对权重.
| S ij |度量相互作用矩阵的稀疏性; λ 控制稀疏和低秩的相对权重.
4) 去背景噪音方法的实验结果及分析
我们在GREMLIN测试集上测试去背景噪音方法的性能,详细实验结果表1、表2所示.从表1和表2中可以看出无论序列间隔取值,无论是局部模型或全局模型的具体方法,LRS的去噪效果都一致地优于APC技术,且对局部模型的改善显著高于全局模型.LRS技术的价值集中体现在对局部模型的显著改进,使其达到和全局模型mfDCA相近的性能.这是自全局模型提出以来首次发现基于局部度量的方法能够达到和全局模型可比的效果,也说明只有当有效地去除背景噪音之后,相关性度量才能提取出更加准确的共进化信息.下面我们将从理论上进一步深入分析LRS优于其他去背景噪音方法的原因,主要基于2个事实:
① APC和SC的等价性
SC认为第一主成分表示相关性的整体一致性,能够刻画由进化偏差引起的背景噪声.第一特征值和对应特征向量元素分别近似为
则背景噪音矩阵元素近似为
 .
.
(28)
从上述分析可以看出SC关于背景噪音的近似与APC的平均乘积校正是等价的,都是秩为1的矩阵.
② LRS是SC和APC的扩展和加强
LRS用低秩矩阵近似背景噪声,是上述2种技术的扩展;另外,用稀疏矩阵表征信号矩阵符合真实残基接触的稀疏性事实.这从理论上保证了LRS方法的优越性 [36] .
我们预期LRS将取代APC成为有效的去除背景噪声的手段.
4.1 基本思想
1) 生物学观察.残基相互作用本身有一定的规律,蛋白质残基的性质,如二级结构、溶液可及性、疏水性等,对残基间形成接触有重要的作用.举例来说,不同的二级结构对于接触的分布有重大影响,如Beta正平行和反平行片段之间的残基接触呈现出完全不同的模式.
2) 数学模型设计.监督学习方法将残基间相互作用预测视为机器学习中的分类问题,首先对每个残基对都提取多种特征(比如保守性、预测的二级结构、溶液可及表面积等),然后在已知残基相互作用情况的集合上训练上述特征的权重.
4.2 典型方法
各类监督学习方法的不同主要体现在2方面:1)选取的特征不同;2)采用的机器学习的模型不同.典型的方法包括整数规划PhyCMAP [37] 、随机森林PconsC系列 [38-39] 和神经网络方法MetaPSICOV [11] ,简要介绍如下.
4.2.1 典型方法1:整数线性规划
Xu等人 [37] 考虑残基性质对残基远程相互作用的重大影响,利用整数线性规划将残基相互作用需满足的物理约束和共进化信息整合起来,开发了软件PhyCMAP.其基本思想是:1)采用随机森林技术预测残基间存在相互作用的概率;2)利用整数线性规划选择出概率较大的残基间相互作用,同时要求这些残基对满足一些物理约束.
整数线性规划的目标函数为
(29)
其中0-1变量 Y i , j 表示残基 i 和 j 之间是否有相互作用; P i , j 为随机森林预测的残基 i 和残基 j 之间存在相互作用的概率; ![]() U r 表示物理约束的惩罚项.
U r 表示物理约束的惩罚项.
整数线性规划考虑以下物理约束:残基 i 最多参与形成多少残基相互作用;2个二级结构之间最多会形成多少残基相互作用;2个strand(形成sheet的片段单元)之间的相互作用具有连续性等.比如当2个strand之间形成正平行sheet时,接触的相邻残基对需要满足以下约束: Y i , j ≥ Y i -1, j + Y i +1, j +1 -1,其中, i , i ±1表示其中一个strand上的残基, j , j ±1表示另一个strand上的残基.该约束保证2个strand之间形成的相互作用具有连续性.
PhyCMAP由于同时考虑了真实残基相互作用的限制和共进化信息,其在CASP10和Set600数据集 [37] 上测试的结果表明PhyCMAP超过了当时比较流行的其他监督学习方法的软件,例如NNcon [12] ,SVMcon [40] 等.
同时PhyCMAP也有其局限性.其只考虑了局部模型MI输出的共进化信息,而并没有考虑更加有效的全局模型输出的共进化信息.下面介绍的Pconsc和MetaPSICOV方法克服了PhyCMAP的局限性.
4.2.2 典型方法2:随机森林方法PconsC系列
Skwark等人 [39] 发现不同的全局模型预测得到的残基接触集合有一定差异;而且不同的构建多序列联配的软件输出的多序列联配也不同,这些不同的多序列联配也会导致远程相互作用的预测结果不同.
基于上述观察,2013年Skwark等人 [39] 将2种全局模型PSICOV和plmDCA对8种不同多序列联配的预测结果与其他特征整合,提出了预测残基间相互作用的随机森林方法,并开发了软件PconsC.多序列联配由HHblits和jackhamme两种比对软件取定4种不同的阈值得到;其考虑的残基对特征包括二级结构预测值、残基溶液可及表面积、残基替代向量.实验结果表明:PconsC具有较高的预测精度,超过PSICOV和plmDCA的预测结果.
基于“相互作用残基对的成簇性”这一认识,Skwark等人 [38] 进一步改进PconsC,用多层随机森林逐步过滤掉孤立的相互作用对,开发了软件PconsC2.值得指出的是,PconsC2的另一个优势在于显著减少了对样本数的要求,从而首次实现当同源序列少于1 000条时的准确预测.实验结果表明:PconsC2比已有的phyCMAP具有更优的预测准确率.
4.2.3 典型方法3:神经网络方法MetaPSICOV
如4.2.1节所述,PhyCMAP结合了结构特征和共进化信息,但是只是引入了局部的共进化信息,并没有引入更加有效的全局共进化信息.Jones等综合考虑结构特征和全局的共进化信息,提出了预测残基间相互作用的神经网络模型,并开发了软件metaPSICOV [11] .
具体地,metaPSICOV是一个2层前向神经网络模型:第1层基于二级结构、溶液可及性、残基替代向量等特征以及PSICOV,mfDCA,plmDCA的预测结果,利用含55个隐单元的神经网络预测出残基相互作用概率的粗略估计;第2层以第1层的粗略估计为输入特征,再加上部分结构特征,使用相同的神经网络对相互作用概率的估计进行校正.
metaPSICOV根据MSA质量比较准确地权衡共进化特征和传统特征(如二级结构等)的权重,从而更加有效地整合多种信息,提高预测准确率.结果显示,metaPSICOV超过PSICOV,mfDCA,plmDCA的预测效果,并在第11届CASP竞赛中获第1名(详细分析见第6节).
4.3 监督学习方法的实验结果及分析
针对监督学习方法的实验结果分析表明2点结论:
1) 早期的监督学习方法,例如采用支持向量机模型的SVMcon [12] 和采用神经网络模型的NNcon [40] 等,由于没有加入有效的共进化特征,其效果并不比无监督学习方法的全局模型好.
2) 近年来提出的监督学习方法metaPSICOV和Pconsc2等,综合了多种结构特征和无监督方法输出的共进化特征,其效果超过了无监督学习方法和早期的监督学习方法,从而表明将无监督方法得到的结果整合到监督学习方法中是当前预测残基相互作用最有效的策略.
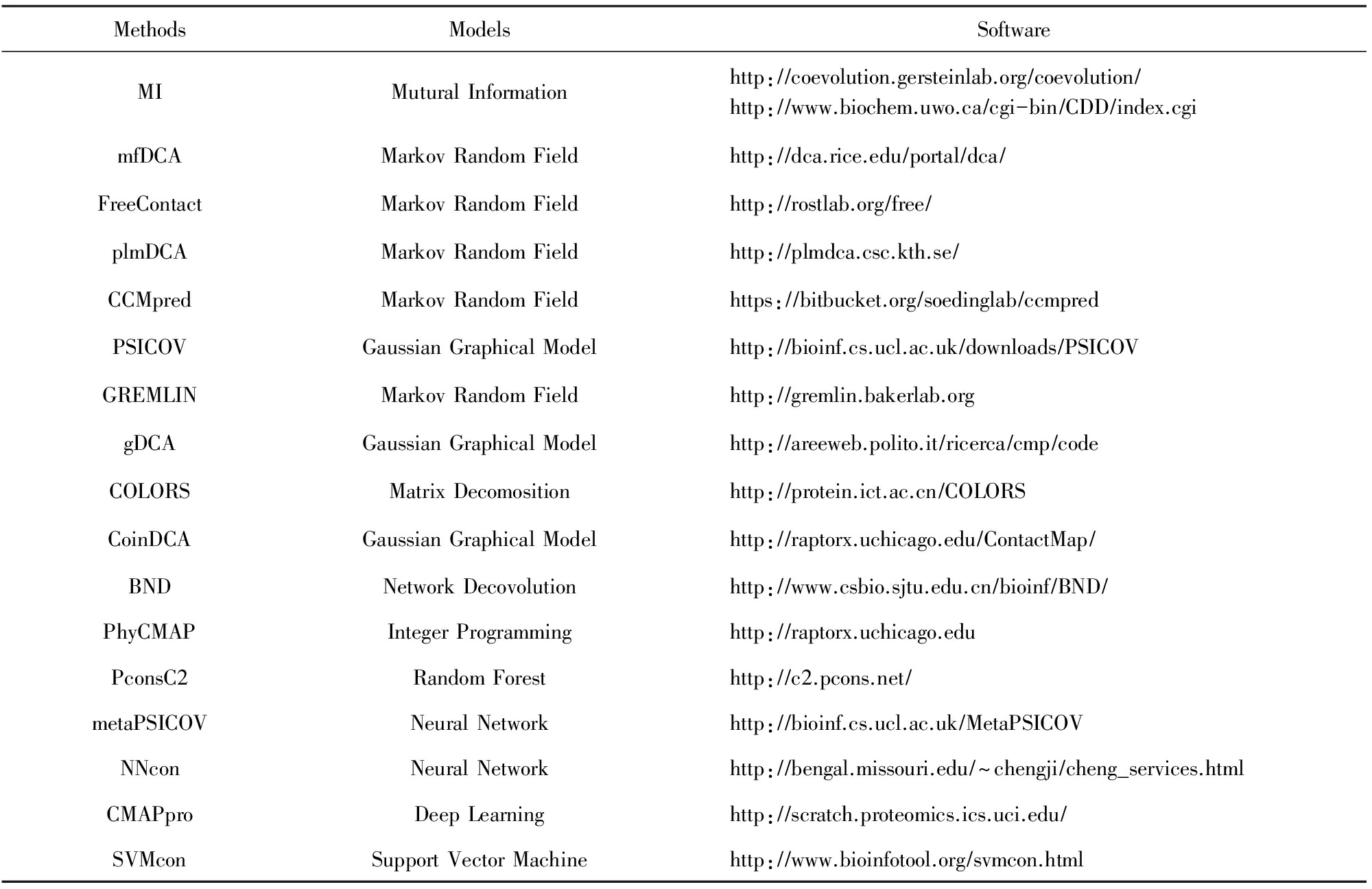
第2.4节所述的方法都有相应的服务器为用户提供残基相互作用预测服务,我们将这些服务器汇总如表3所示:
Table 3 Overview of Existing Softwares for Protein Contact Prediction
表3 现有残基相互作用预测软件概览
CASP竞赛是全球范围内的蛋白质结构预测比赛,现已作为客观评估蛋白质结构预测质量的标准.从1994年开始,每两年1届,迄今已举办11届.目前,CASP竞赛包括结构预测、残基远程相互作用预测、接触位点辅助结构预测、结构优化、结构质量评估5个部分.
在蛋白质结构预测领域,大部分软件采用开源软件或者免费预测服务的方式,商业软件较少(比如DNAStar公司开发的NovaFold和BSI公司开发的RAPTOR等).国内研究团队也多次参加CASP比赛,包括中科院生物物理研究所的Jiang-Server团队、上海交通大学的Shen-group团队以及中科院计算所的FALCON团队.其中本课题团队开发的FALCON系列软件在CASP-8中结构预测FR-Hard类上获得第3名,在CASP-11中结构预测TBM类上获得第9名.Shen-group在CASP-11残基接触预测的FM类蛋白上取得了第2名(以precision评价).
残基远程相互作用预测作为CASP竞赛的重要部分.在2014年的第11届CASP比赛中,共有29个软件参加了残基相互作用预测 [41-45] .本文提到的一些经典算法参加了这次比赛,例如采用PhyCMAP方法的RaptorX-Contact软件、采用MetaPSICOV的CONSIP2软件以及采用PconsC2方法的MetaPSICOV软件等.
在CASP11中,参赛软件大多是基于监督学习的方法.在监督学习方法中,无监督学习全局模型输出的信息是其重要特征.根据是否含有全局共进化模型信息,可以将这些监督学习方法分为2类:1)不包含全局共进化模型信息的方法:例如PhyCMAP,采用SVMcon方法的MULTICOM-construct、采用DNcon方法的MULTICOM-cluster软件和采用NNcon方法的MULTICOM-novel软件等;2)包含全局共进化模型信息的方法:例如采用神经网络模型的CONSIP2方法、采用随机森林模型的Pcons-net(PconsC2)、采用SVM模型的Shen-group和RBO_ALEPH [46] 等.我们的实验结果分析表明包含全局共进化模型信息的方法要好于不包含全局共进化模型信息的方法.
CASP竞赛对于方法的衡量主要有4个分项:
1) 预测准确率( precision )
根据各方法输出的分数进行排序,选取Top N 作为预测的接触集合,其他的作为非接触集合.其中 N 一般取 L 10, L 5, L 2或 L 等( L 为目标蛋白的序列长度),计算正阳性(true positive, TP )和假阳性(false positive, FP ), precision 的定义为
![]() .
.
2) Xd 值
将氨基酸对之间的距离分成15个区间:(0,4]Å,…,(56,60]Å. Xd 值的定义为
其中, Pp i 表示预测的残基接触的距离在第 i 个区间的比例, Pa i 表示结构所有的残基接触对的距离在 i 个区间的比例. Xd 用来衡量真实结构和预测接触中残基对距离分布的差异.
3) Matthews相关系数( MCC )
CASP比赛要求各参赛软件给出每个残基对接触的概率.选取0.5作为阈值计算 TP , TN , FP (false positive)和 FN (false negative).
MCC =( TP × TN - FP × FN )
![]() .
.
4) precision-recall曲线下的面积( AUC_PR )
由于上述共进化分析方法给出相关性度量,而非残基对的接触概率,Matthews相关系数并不适用于这些方法,所以我们采用 precision , Xd 值和 AUC_PR 这3种分项对这些方法进行评估.
我们在CASP11比赛数据集合测试了本文综述的典型方法以及参加CASP比赛的主要方法,对实验结果采用自行开发的程序进行了详细分析(程序和数据下载地址http: 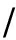 bioinfo.ict.ac.cn COLORS ),详细分析结果按照无模板建模(free modelling, FM)类和有模板建模(template based modelling, TBM)类分别描述如下:
bioinfo.ict.ac.cn COLORS ),详细分析结果按照无模板建模(free modelling, FM)类和有模板建模(template based modelling, TBM)类分别描述如下:
6.1 在FM类目标蛋白上的测试结果
在CASP11比赛中,共有17参赛队伍提交了超过20个FM类蛋白质域的预测结果.图10列出了各方法的预测性能,包括 precision , Xd 和 AUC_PR .从图10我们可以得到如下结论.
1) 综合全局共进化模型信息的监督方法领先于其他方法.比如CONSIP(metaPSICOV)在3种分项都取得第1名;Shen-group也处于领先地位.以 precision 和 AUC_PR 评价,RBO_ALEPH的排名也较靠前.
2) 监督学习方法整体上优于无监督学习方法的效果.例如以 precision 作评价,无论是包含了全局共进化信息的监督方法(如CONSIP,Shen-group)还是不包含全局共进化信息的监督方法(如MULTICOM-novel,RaptorX,MULTICOM-cluster),都超过了无监督学习的方法(如plmDCA,PSICOV).
3) 在无监督学习方法中,全局模型普遍优于无监督学习方法.
4) 以 precision 作为评价,LRS技术优于APC技术.
值得指出的是:不同预测方法在不同的评价指标下表现不同,例如RaptorX以 precision 为评价排名为16,而以 Xd 为评价排名为2;对于无监督学习方法的去噪音方法,以 precision 为评价,LRS好于APC;但以 AUC_PR 为评价,APC好于LRS.其原因在于:LRS只提取显著的共进化信号,将非显著的共进化信号的分数设置为0;而APC却可以对非显著的共进化信号进行排名,从而造成如果以 AUC_PR 为评价,APC技术优于LRS技术.
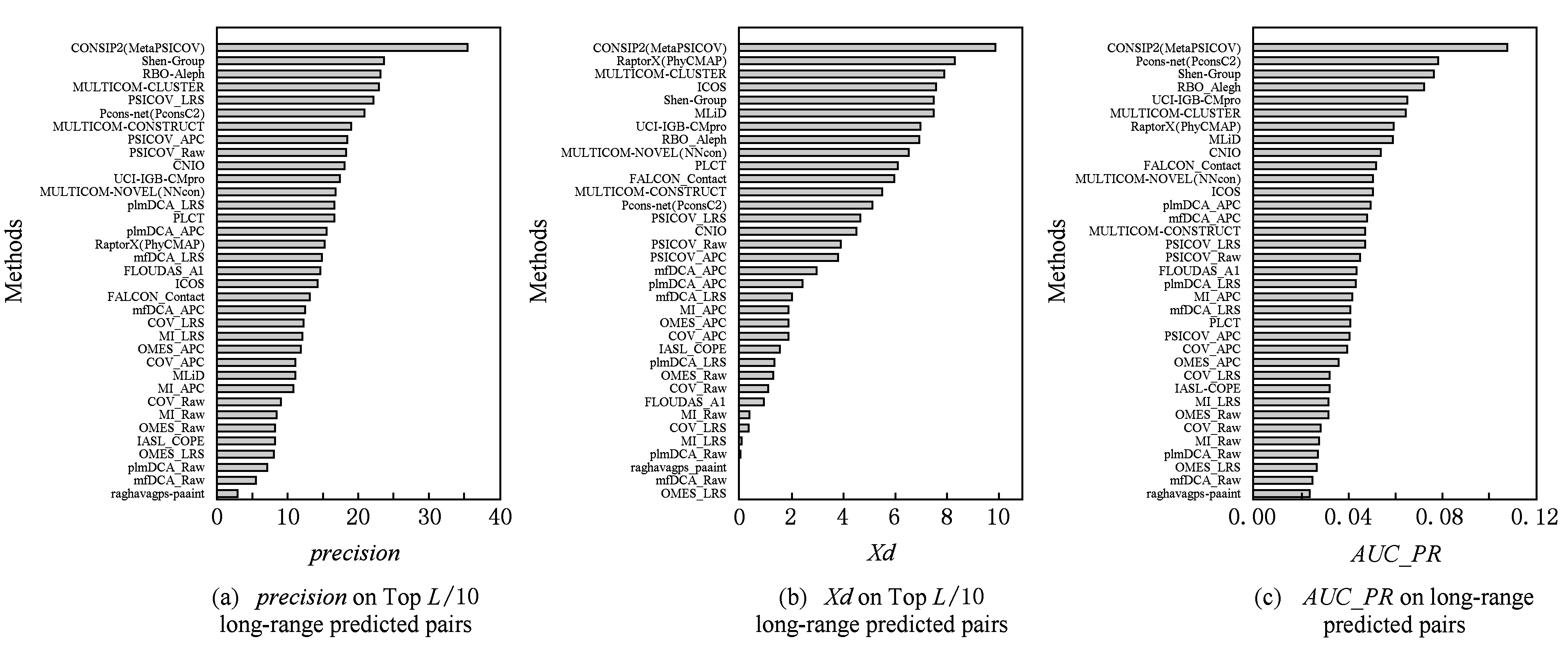
Fig. 10 Prediction performance of different methods on CASP-11 FM targets
图10 典型方法对CASP-11 FM类蛋白的预测性能
6.2 在TBM类目标蛋白上的测试结果
在CASP11比赛中,共有14个参赛组提交了多于60个TBM类别蛋白域的预测结果,如图11所示:
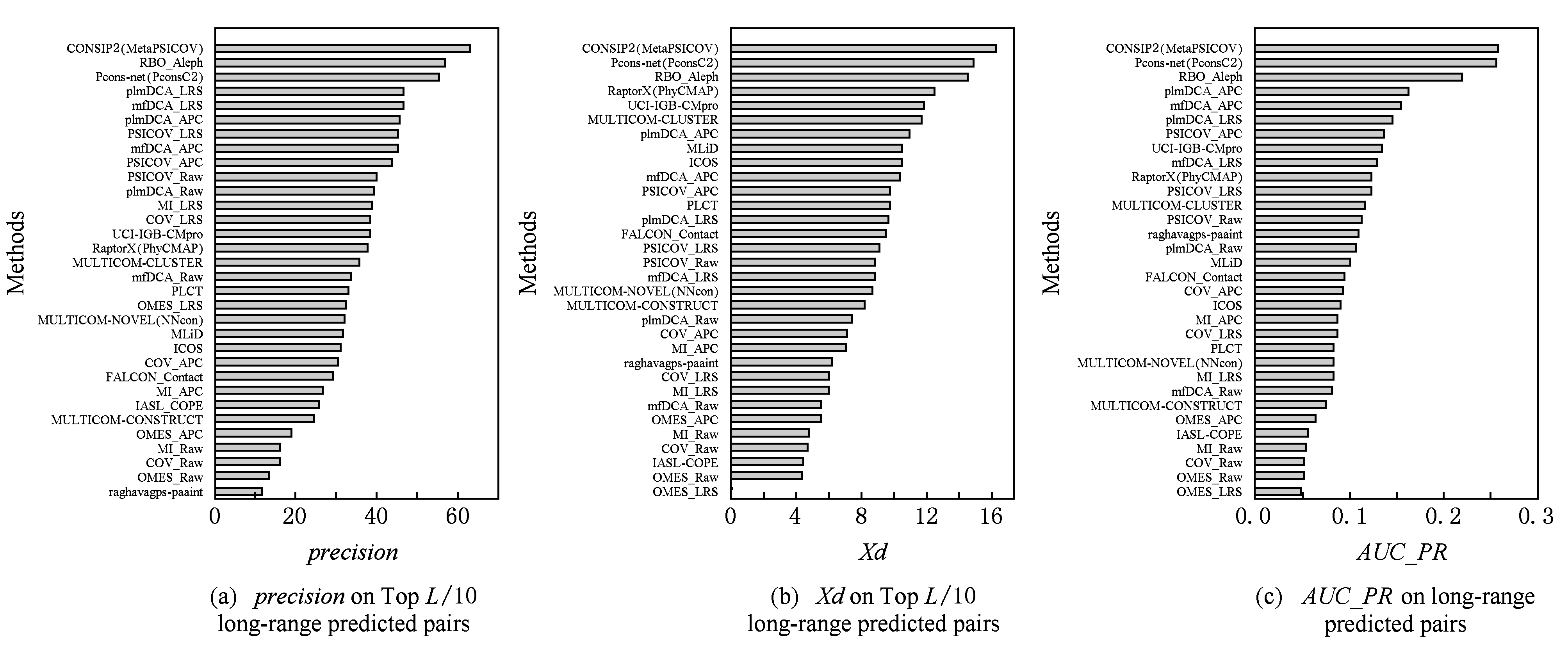
Fig. 11 Prediction performance of different methods on CASP-11 TBM targets
图11 典型方法对CASP-11 TBM类蛋白的预测性能
我们可得到如下结论:
1) 与FM类目标蛋白上的观察相同,综合了全局共进化模型信息的监督方法领先于其他方法.
2) 以 precision 为评价,整体上来讲,无监督学习方法中的全局模型优于没有结合全局模型信息的监督学习方法.例如,plmDCA_LRS和PSICOV_LRS的预测性能优于RaptorX和MULTICOM-cluster.这在很大程度上源于TBM目标蛋白的多序列联配比FM包含更多的同源序列,从而提供了更准确的共进化信息.
3) 与FM类目标蛋白上的观察相同,无监督学习方法中的全局模型整体优于局部模型.
6.3 预测准确率与有效同源序列数目的关系
多序列联配的有效同源序列的数目对预测准确率的影响很大.无监督学习方法中性能较好的全局模型往往具有较多的参数,从而需要大量的同源序列进行参数估计;在监督学习中使用的重要特征也受同源序列数目的影响,例如序列谱、预测的二级结构和预测的可及水表面积等特征.
我们在99个FM类和TBM类目标蛋白上测试同源序列数目对各方法预测准确度的影响,选取经典的无监督学习方法和在CASP11中提交超过90个蛋白域的软件进行评价.
给定多序列联配,我们采用了通常使用的有效序列数的定 ![]() w k .其中 w k 为多序列联配中第 k 条序列的数目的权重.
w k .其中 w k 为多序列联配中第 k 条序列的数目的权重.
我们根据 Meff 将这些目标蛋白分成了3组:1)(0,100],共36个蛋白;2)(100,1 000],共32个蛋白;3) Meff >1 000,共31个蛋白.
如图12所示,我们可以得到以下结论:
1) 所有方法的预测准确度都随 Meff 的提高而提高,但不同方法的提高程度不同.
2) 总体来讲,融合全局度量的监督学习方法在3种 Meff 类别下都领先于其他方法,例如CONSIP2(MetaPSICOV),Pcons-net(Pcons2),RBO_Aleph等.
3) 当 Meff 较低时,不融合全局度量的监督学习方法优于无监督方法的全局模型.例如MULTICOM-CONSTRUCT,RaptorX-Conact,MULTICOM_CLUSTER优于CCMpred,PSICOV等方法.
4 当 Meff 较高时,全局模型逐渐超越不含全局模型信息的无监督方法.
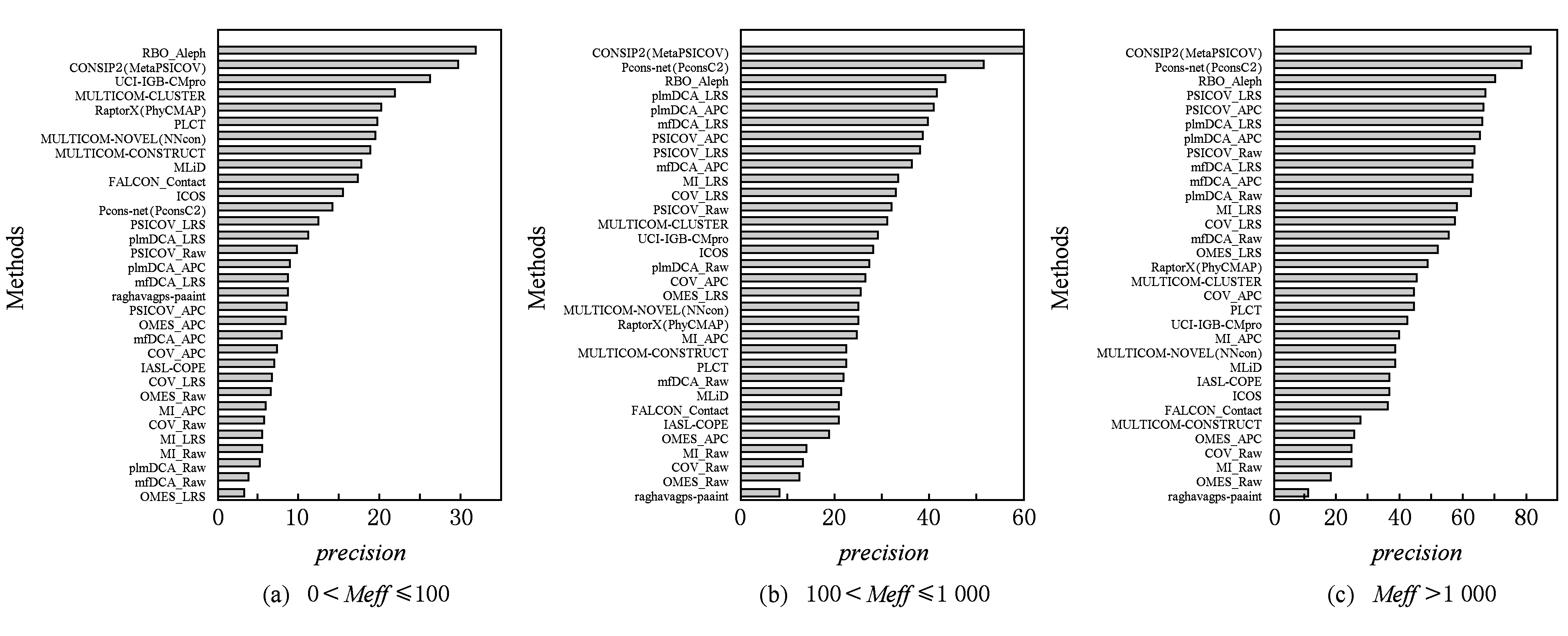
Fig. 12 Relationship of mean precision with Meff for different methods on CASP-11 targets(FM and TBM)
图12 典型方法对CASP-11 (FM和TBM)蛋白预测的平均准确率与Meff的关系
残基共进化分析可在未知蛋白质结构时,仅依据序列推断残基间的相互作用,因此在蛋白质结构和功能预测上具有重要的研究意义.
1) 残基间远程相互作用的信息能够有效地促进对蛋白质结构的预测,其典型工作是基于mfDCA的相互作用预测信息开发的蛋白质结构预测软件DCA-fold [47] 和EVfold [14] .Hopf等人 [48] 考虑膜蛋白的结构特性,使用EVfold对DrugBank数据库中23个已知结构的膜蛋白家族进行结构预测,发现其中20个可以得到高精度预测,进而对Pfam中11个未知结构蛋白质进行结构预测.
2) 由于蛋白质结构中的二硫键可以看成一种特殊的残基相互作用,所以相互作用预测的信息也有助于二硫键的预测.Yang等人提取远程相互作用预测软件GREMLIN的输出信息,作为支持向量回归(support vector regression, SVR)模型的特征之一 [49] .结果表明其软件Cyscon优于其他同类软件.
3) 残基间相互作用能够有助于对残基功能的推断.一般地,功能位点倾向于是蛋白质中的保守位点;类似地,残基间的关联强度也有助于推断功能位点,比如Marks等人 [6,48] 计算某特定残基与其他残基的累积耦合强度,作为该残基功能性(功能选择压力)的度量,并发现了一些重要的功能元件.
经过20多年的发展,研究者已经提出了多种残基相互作用的预测模型,使得预测精度有了显著提升.然而目前已有的算法依然存在假阳性率较高、对样本数目显著依赖等缺陷.在前期工作的基础上,我们认为残基间相互作用有3个发展趋势:
趋势1. 改进参数估计方法.经典的统计模型如Markov随机场能够比较准确地描述蛋白质所有位点的全概率分布,但其参数估计的各种近似策略还有待改善,以进一步缩小与极大似然估计之间的差异,并提高计算效率.
趋势2. 预测二级结构单元之间的相互作用.传统远程相互作用预测方法都是预测残基间相互作用,这在计算上具有很大便利,但是同时也造成显著的假阳性.事实上,从预测蛋白质整体结构这个目标来说,判断二级结构单元之间是否存在相互作用就能够提供足够有价值的信息,而不用细化到残基间是否存在相互作用.
趋势3. 改进相互作用预测的评价方法.目前的评价方法中,所有残基对是同等考虑的.然而在蛋白质中,各个残基的重要性是有显著差异的,比如二级结构单元之间的相互作用、疏水集团与其他位点的相互作用等具有相对更高的重要性,而突变较多较随机的不重要的位点则对结构影响不大.一种可能的方案是首先基于多序列联配给出位点重要性的先验概率,进而在相互作用预测中有侧重地考虑那些重要的残基,这种有针对性地设置约束将能够提高结构预测效率和精度.
本文对残基间远程相互作用预测进行了综述,主要介绍了残基间相互作用预测的机器学习方法,分析了各方法的预测性能,并总结了未来的发展趋势.
值得指出的是,残基间相互作用预测是机器学习中结构学习(structured learning)的一个典型问题,因此这方面的研究不仅具有重要的生物学意义,同时能够推动机器学习领域的研究.
参考文献:
[1]Lodish H F, Berk A, Zipursky S L, et al. Molecular Cell Biology[M]. New York: WH Freeman, 2000
[2]Petsko G A, Ringe D. Protein Structure and Function[M]. London: New Science Press, 2004
[3]Wüthrich K. The way to NMR structures of proteins[J]. Nature Structural & Molecular Biology, 2001, 8(11): 923-925
[4]Kendrew J C, Bodo G, Dintzis H M, et al. A three-dimensional model of the myoglobin molecule obtained by X-ray analysis[J]. Nature, 1958, 181(4610): 662-666
[5]Taylor K A, Glaeser R M. Electron diffraction of frozen, hydrated protein crystals[J]. Science, 1974, 186(4168): 1036-1037
[6]Marks D S, Hopf T A, Sander C. Protein structure prediction from sequence variation[J]. Nature Biotechnology, 2012, 30(11): 1072-1080
[7]Anfinsen C B. Principles that govern the folding of protein chains[J]. Science, 1973, 181(4096): 223-230
[8]Kim De, Dimaio F, Wang R Y, et al. One contact for every twelve residues allows robust and accurate topology-level protein structure modeling[J]. Proteins: Structure, Function, and Bioinformatics, 2014, 82(S2): 208-218
[9]Haile J M. Molecular Dynamics Simulation[M]. New York: Wiley, 1992
[10]Anzai Y. Pattern Recognition and Machine Learning[M]. New York: Academic Press, 2012
[11]Jones D T, Singh T, Kosciolek T, et al. MetaPSICOV: Combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins[J]. Bioinformatics, 2015, 31(7): 999-1006
[12]Cheng Jianlin, Baldi P. Improved residue contact prediction using support vector machines and a large feature set[J]. BMC Bioinformatics, 2007, 8(1): 11-13
[13]Chen Peng. Analysis and prediction of interactions between residues in proteins[D]. Hefei: University of Science and Technology of China, 2007 (in Chinese)(陈鹏. 蛋白质残基间的相互作用分析与预测[D]. 合肥: 中国科学技术大学, 2007)
[14]Marks D S, Colwell L J, Sheridan R, et al. Protein 3D structure computed from evolutionary sequence variation[J]. PLoS ONE, 2011, 6(12): 1287-1296
[15]Gobel U, Sander C, Schneider R, et al. Correlated mutations and residue contacts in proteins[J]. Proteins: Structure, Function and Bioinfomatics, 1994, 18(4): 309-317
[16]Martin L C, Gloor G B, Dunn S D, et al. Using information theory to search for co-evolving residues in proteins[J]. Bioinformatics, 2005, 21(22): 4116-4124
[17]Kass I, Horovitz A. Mapping pathways of allosteric communication in GroEL by analysis of correlated mutations[J]. Proteins: Structure, Function, and Bioinformatics, 2002, 48(4): 611-617
[18]Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence-and structure-rich era[J]. Proceedings of the National Academy of Sciences, 2013, 110(39): 15674-15679
[19]Morcos F, Pagnani A, Lunt B, et al. Direct-coupling analysis of residue coevolution captures native contacts across many protein families[J]. Proceedings of the National Academy of Sciences, 2011, 108(49): 1293-1301
[20]Weigt M, White R A, Szurmant H, et al. Identification of direct residue contacts in protein-protein interaction by message passing[J]. Proceedings of the National Academy of Sciences, 2009, 106(1): 67-72
[21]Lövkvist C, Lan Y, Weigt M, et al. Improved contact prediction in proteins: Using pseudolikelihoods to infer Potts models[J]. Physical Review E, 2013, 87(1): 12707-12929
[22]Burger L, van Nimwegen E. Disentangling direct from indirect co-evolution of residues in protein alignments[J]. PLoS Computational Biology, 2010, 6(1): 10006-10033
[23]Ma Jianzhu, Wang Sheng, Wang Zhiyong, et al. Protein contact prediction by integrating joint evolutionary coupling analysis and supervised learning[J]. Bioinformatics, 2015, 31(21): 3506-3513
[24]Jones D T, Buchan D W, Cozzetto D, et al. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments[J]. Bioinformatics, 2012, 28(2): 184-190
[25]Feizi S, Marbach D, Médard M, et al. Network deconvolution as a general method to distinguish direct dependencies in networks[J]. Nature biotechnology, 2013, 31(8): 726-733
[26]Meilă M, Jaakkola T. Tractable Bayesian learning of tree belief networks[C]  Proc of the 6th Conf on Uncertainty in Artificial Intelligence. San Francisco, CA: Morgan Kaufmann, 2000: 380-388
Proc of the 6th Conf on Uncertainty in Artificial Intelligence. San Francisco, CA: Morgan Kaufmann, 2000: 380-388
[27]Lapedes A S, Bertrand G G, Liu L, et al. Correlated mutations in models of protein sequences: Phylogenetic and structural effects[J]. Lecture Notes-Monograph Series, 1999, 33(1), 236-256
[28]Plefka T. Convergence condition of the TAP equation for the infinite-ranged Ising spin glass model[J]. Journal of Physics A: Mathematical and general, 1982, 15(6): 1971-1985
[29]Georges A, Yedidia J S. How to expand around mean-field thery using high-temperature expansions[J]. Journal of Physics A: Mathematical and General, 1991, 24(9): 2173-2179
[30]Csisz X, R I, Talata Z. Consistent estimation of the basic neighborhood of Markov random fields[J]. The Annals of Statistics, 2006, 34(1): 123-145
[31]Lauritzen S L. Graphical Models[M]. Oxford, UK: Oxford University Press, 1996
[32]Wright S. Correlation and causation[J]. Journal of Agricultural Research, 1921, 20(7): 557-585
[33]Sun Haiping, Huang Yan, Wang Xiaofan, et al. Improving accuracy of protein contact prediction using balanced network deconvolution[J]. Proteins: Structure, Function, and Bioinformatics, 2015, 83(3): 485-496
[34]Dunn S D, Wahl L M, Gloor G B. Mutual information without the influence of phylogeny or entropy dramatically improves residue contact prediction[J]. Bioinformatics, 2008, 24(3): 333-340
[35]Halabi N, Rivoire O, Leibler S, et al. Protein sectors: Evolutionary units of three-dimensional structure[J]. Cell, 2009, 138(4): 774-786
[36]Zhang Haicang, Gao Yujuan, Deng Minghua, et al. Improving residue-residue contact prediction via low rank and sparse decomposition of residue correlation matrix[J]. Biochemical and Biophysical Research Communications, 2016, 472(1): 217-222
[37]Wang Zhiyong, Xu Jinbo. Predicting protein contact map using evolutionary and physical constraints by integer programming[J]. Bioinformatics, 2013, 29(13): 266-273
[38]Skwark M J, Raimondi D, Michel M, et al. Improved contact predictions using the recognition of protein like contact patterns[J]. PLoS Computatioal Biology, 2014, 10(11): 1003-1019
[39]Skwark M J, Abdel-Rehim A, Elofsson A. PconsC: Combination of direct information methods and alignments improves contact prediction[J]. Bioinformatics, 2013, 29(14): 1815-1816
[40]Tegge A N, Wang Z, Eickholt J, et al. NNcon: Improved protein contact map prediction using 2D-recursive neural networks[J]. Nucleic Acids Research, 2009, 37(Suppl 2): 515-518
[41]Kajan L, Hopf T A, Kalas M, et al. FreeContact: Fast and free software for protein contact prediction from residue co-evolution[J]. BMC Bioinformatics, 2014, 15(1): 158-164
[42]Seemayer S, Gruber M, Söding J. CCMpred-fast and precise prediction of protein residue-residue contacts from correlated mutations[J]. Bioinformatics, 2014, 30(21): 3128-3130
[43]Baldassi C, Zamparo M, Feinauer C, et al. Fast and accurate multivariate Gaussian modeling of protein families: Predicting residue contacts and protein-interaction partners[J]. PLoS ONE, 2014, 9(3): 927-940
[44]Di Lena P, Nagata K, Baldi P. Deep architectures for protein contact map prediction[J]. Bioinformatics, 2012, 28(19): 2449-2457
[45]Monastyrskyy B, D’Andrea D, Fidelis K, et al. New encouraging developments in contact prediction: Assessment of the CASP11 results[J]. Proteins: Structure, Function, and Bioinformatics, 2015, 6(4): 126-140
[46]Schneider M, Brock O. Combining physicochemical and evolutionary information for protein contact prediction[J]. PloS ONE, 2014, 9(10): 1108-1120
[47]Su  kowska J I, Morcos F, Weigt M, et al. Genomics-aided structure prediction[J]. Proceedings of the National Academy of Sciences, 2012, 109(26): 10340-10345
kowska J I, Morcos F, Weigt M, et al. Genomics-aided structure prediction[J]. Proceedings of the National Academy of Sciences, 2012, 109(26): 10340-10345
[48]Hopf T A, Colwell L J, Sheridan R, et al. Three-dimensional structures of membrane proteins from genomic sequencing[J]. Cell, 2012, 149(7): 1607-1621
[49]Yang Jing, He Baoji, Jang R, et al. Accurate disulfide-bonding network predictions improve ab initio structure prediction of cysteine-rich proteins[J]. Bioinformatics, 2015, 31(23): 3773-3781

Zhang Haicang, born in 1987. PhD candidate from the Institute of Computing Technology, Chinese Academy of Sciences. His main research interests include bioinformatics, algorithm design and machine learning.

Gao Yujuan, born in 1992. PhD candidate from Peking University. Her main research interests include network inference and convex optimization algorithm.

Deng Minghua, born in 1969. Received his BS, MS, and PhD degrees in applied mathematics from Peking University. Professor in the School of Mathematical Sciences, Centre for Quantitative Biology and Center for Statistical Sciences, Peking University. His main research interests include bioinformatics and system biology.

Zheng Weimou, born in 1946. Received his BS degree in the Department of Physics from Peking University and PhD degree from Universite Libre de Bruxelles. Professor of the Institute of Theoretical Physics, Chinese Academy of Sciences. His main research interests include surface physics, stochastic process, nonlinear dynamics, biophysics and bioinformatics.

Bu Dongbo, born in 1973. Received his BS in computer science, MS and PhD degrees from the Institute of Computing Technology, Chinese Academy of Sciences. Professor of the Institute of Computing Technology, Chinese Academy of Sciences. Member of CCF. His main research interests include algorithm design and analysis, SAT problem, and bioinformatics (especially on genome sequencing assembly, protein sequencing via mass spectra, protein structure prediction).
Zhang Haicang 1,2 , Gao Yujuan 3 , Deng Minghua 3,4,5 , Zheng Weimou 6 , and Bu Dongbo 1 1
( Institute of Computing Technology , Chinese Academy of Sciences , Beijing 100190) 2 ( University of Chinese Academy of Sciences , Beijing 100049) 3 ( Centre for Quantitative Biology , Peking University , Beijing 100871) 4 ( School of Mathematical Sciences , Peking University , Beijing 100871) 5 ( Center for Statistical Sciences , Peking University , Beijing 100871) 6 ( Institute of Theoretical Physics , Chinese Academy of Sciences , Beijing 100190)
Abstract: Proteins are large molecules consisting of a linear sequence of amino acids. In the natural environment, a protein spontaneously folds into specific tertiary structure to perform its biological functionality. The main factors that drive proteins to fold are interactions between residues, including hydrophobic interaction, Van der Waals’ force and electrostatic interactions. The interactions between residues usually lead to residue-residue contacts, and the prediction of residue-residue contacts should greatly facilitate understanding of protein structures and functionalities. A great variety of techniques have been proposed for residue-residue contacts prediction, including machine learning, statistical models, and linear programing. It should be pointed out that most of these techniques are based on the biological insight of co-evolution, i.e., during the evolutionary history of proteins, a residue’s mutation usually leads its contacting partner to mutate accordingly. In this review, we summarize the state-of-art algorithms in this field with emphasis on the construction of statistical models based on biological insights. We also present the evaluation of these algorithms using CASP (critical assessment of techniques for protein structure prediction) targets as well as popular benchmark datasets, and describe the trends in the field of protein contact prediction.
Key words: protein contact prediction; protein tertiary structure prediction; graphical model; co-evolution; machine learning
收稿日期: 2015-12-10;
修回日期: 2016-04-14
基金项目: 国家“九七三”重点基础研究发展计划基金项目(2012CB316502,2015CB910303);国家自然科学基金项目(11175224,11121403,31270834,61272318,31171262,31428012,31471246);中国科学院理论物理研究所理论物理国家重点实验室开放工程项目(Y4KF171CJ1) This work was supported by the National Basic Research Program of China (973 Program) (2012CB316502, 2015CB910303), the National Natural Science Foundation of China (11175224, 11121403, 31270834, 61272318, 31171262, 31428012, 31471246), and the Open Project Program of State Key Laboratory of the Institute of Theoretical Physics, Chinese Academy of Sciences (Y4KF171CJ1).
通信作者: 高玉娟(lacus2009@163.com,其对本文的贡献同第一作者);卜东波(dbu@ict.ac.cn)
中图法分类号: TP399