吴建盛 1 冯巧遇 2 袁京洲 1 胡海峰 2 周家特 1 高 昊 1
1 (南京邮电大学地理与生物信息学院 南京 210023) 2 (南京邮电大学通信与信息工程学院 南京 210003) (jansen@njupt.edu.cn)
摘 要 G蛋白偶联受体(G protein-coupled receptors, GPCRs)是人类中最庞大的膜蛋白家族,也是很多药物的重要靶点,准确了解GPCRs生物学功能是理解它们参与的生物学过程及其药物作用机制的关键.以前的研究表明,蛋白质功能预测可抽象为多示例多标记学习(multi-instance multi-label learning, MIML)问题.设计了一种基于快速多示例多标记学习方法MIMLfast的GPCRs生物学功能预测模型.该模型采用了一种新的混合特征,它考虑了GPCRs结构域的三联氨基酸、氨基酸关联、进化、二级结构关联、信号肽及无序残基等多种信息.实验结果证明,该模型获得了很好的性能,优于目前最优的多示例多标记学习、多标记学习的预测方法和CAFA蛋白质功能预测方法.
关键词 G蛋白偶联受体;生物学功能预测;快速多示例多标记学习;结构域;混合特征
G蛋白偶联受体(G protein-coupled receptors,GPCRs)具有7个跨膜结构域,是细胞信号传导过程中的重要蛋白质,它在人类视觉、嗅觉、味觉、神经传递、新陈代谢以及免疫调节等各项正常生理活动和疾病过程中都发挥着重要的作用 [1] .另外,大约40%的现代药物都以GPCRs作为靶点 [2] .准确了解GPCRs的生物学功能是理解它们参与的生物学过程及其药物作用机制的关键.
蛋白质通常含有多个保守结构域,同时具有多种生物学功能,每个结构域可以独立或者与相邻结构域相互作用执行生物学功能.我们以前的研究发现,蛋白质功能预测可抽象为多示例多标记学习(multi-instance multi-label learning, MIML)问题,每个蛋白质对应于多示例多标记学习中的一个样本对象,每个结构域对应于一个示例,每个功能对应于一个标记 [3] .
Zhang & Zhou(2006)提出了多示例多标记学习(MIML)框架 [4] .在该学习框架下,每个训练样本由多个示例表示且同时隶属于多个概念标记.近年来,多示例多标记学习模型由于其强大的表示能力得到了研究者的广泛关注,各种算法被提了出来.其中的代表有基于退化框架下的算法MIMLSVM [4] ,MIMLBOOST [4] ,MIMLNN [5] 和SISL-MIML [6] ;基于正则化机制的算法D-MIMLSVM算法 [5] 和M3MIML算法 [7] ;将单示例数据恢复成MIML形式数据的方法INSDIF [8] ;生成式方法DBA [9] ;MIML距离度量学习 [10] ;MIML示例预测算法Rankloss-Sim [11] .
在过去的研究中,有很多的计算学方法用来预测蛋白质生物学功能 [12] .在这些预测方法中,用于表征蛋白质的属性很多,有氨基酸序列信息、进化信息、基因组分信息、蛋白质-蛋白质相互作用信息、蛋白质结构信息、基因芯片信息等 [12] .蛋白质的生物学功能有多种描述方式,其中基因本体学(gene ontology, GO)使用最为广泛 [13] .基因本体学从3个方面来描述蛋白质的生物学功能,即分子功能(molecular function)、生物学过程(biological process)和细胞组分(cellular component) [13] .GPCRs一般都位于细胞膜上,因此,本文中我们只考虑GPCRs的分子功能和生物学过程.
本文中,我们利用快速多示例多标记学习方法MIMLfast [14] ,基于新的混合特征,对GPCRs的GO分子功能和生物学过程进行了预测.实验结果表明,本文的模型取得了很好的预测性能.
本文先从UniProt数据库 [15] 中下载7tmrlist文件,该文件含有3 052个GPCRs的UniProt ID号,我们通过这些ID号得到所有GPCRs的氨基酸序列.然后,使用NCBI的Blastclust程序 [16] 对GPCRs氨基酸序列进行去冗余处理(相似性小于90%), 通过保留聚类结果中每行的第1个蛋白质得到非冗余的GPCRs样本数据集.
我们从UniProt-GOA ftp站点 [15] 下载得到gene_association.goa_ref_uniprot文件,并通过上面非冗余GPCRs数据集的UniProt ID号得到其GO术语(剔除Evidence Code 为IEA的GO术语).然后,我们从基因本体学数据库 [13] 下载go.obo文件,并运行数据库提供的obo2csv.py程序,得到go.obo.F.is_a和go.obo.P.is_a文件.文件分别提供了每个分子功能和生物学过程GO术语的父节点,这样可以得到GPCRs样本含有的GO术语及其所有父节点GO术语,并得到GPCRs数据集的分子功能和生物学过程的GO术语标记空间.
本文中,我们删除样本个数特别多(大于1 000)和特别少 (小于4)的GO术语;对分子功能,得到非冗余GPCRs样本1 327个,GO术语206个;对生物学过程,得到非冗余GPCRs样本1 331个,GO术语1 406个.
将非冗余GPCRs氨基酸序列文件提交到NCBI的Batch CD-Search服务器 [17] ,得到GPCRs蛋白质的保守结构域.对于每个结构域,本文考虑以下7个特征信息:三联氨基酸信息、氨基酸关联信息、进化信息、二级结构关联信息、信号肽信息、无序残基信息和物化属性.
1) 三联氨基酸信息.本文中,依据氨基酸的偶极矩和侧链体积,把20种氨基酸分为6类:① A 类.有Ala,Gly,Val.② B 类.有Ile,Leu,Phe,Pro.③ C 类.有Tyr,Met,Thr,Ser,Cys.④ D 类.有His,Asn,Gln,Tpr.⑤ E 类.有Arg,Lys.⑥ F 类.有Asp,Glu [18-19] .对于每个结构域,根据其氨基酸序列计算三联体出现频率( Triad ) [19] ,
![]()
(1)
其中, a , b , c ∈{ A , B , C , D , F }; N a bc 表示三联氨基酸的个数; n 为氨基酸序列长度.对每个结构域, Triad 特征为216维.
2) 氨基酸关联信息.依据上面的6类氨基酸 A , B , C , D , E , F ,氨基酸关联信息(amino acid correla-tion, AAC) [20] 为
![]()
(2)
其中, i , j ∈{ A , B , C , D , E , F }; P i 和 P j 分别表示第 i 和 j 类氨基酸在结构域中出现的频率; P i j ( k )表示第 i 和 j 类氨基酸在结构域中间隔 k 个残基的联合出现频率, k ∈{2,4,8,16}.对每个结构域,其AAC特征的维数为144.
3) 进化信息.本文中,位置特异性得分矩阵(position-specific scoring matrics, PSSMs)被用来描述结构域的进化信息.我们通过psiblast程序 [16] 比对NCBI非冗余蛋白质数据集 nr 得到PSSMs矩阵(3轮迭代和期望值设为0.001).对每个结构域,其产生的PSSMs矩阵包含42 n 个元素,其中 n 为氨基酸序列长度. 然后,我们通过标准的logistic函数将PSSMs中的元素 a 归一化到0~1之间,表达式为
![]()
(3)
因为结构域的氨基酸序列长度往往不一样,其得到的PSSMs元素个数将不同.本文中,我们把每个氨基酸残基对应的42个PSSMs元素当着一个示例,每个结构域可表示为由 n 个示例组成的包,其中 n 为结构域的氨基酸序列长度.我们通过多示例算法miFV [21] 将示例包转化为单个向量.对每个结构域,最终得到的PSSMs特征维数为84.
4) 二级结构关联信息.本文中,通 过PSIPRED在线分析工具 [22] 完成结构域的二级结构预测.这里,二级结构包括3类:螺旋(helix)、折叠(sheet)和转角(coil).然后,我们计算了结构域中二级结构关联信息(secondary structure element correlation, SSC) [20] ,
![]()
(4)
其中, i , j ∈{helix,sheet,coil}; P i 和 P j 分别表示第 i 和 j 种二级结构元素在结构域中出现的频率; P i j ( k )表示第 i 和 j 种二级结构元素间隔 k 个残基的联合出现频率, k ∈{2,4,8,16}.对每个结构域,其SSC特征的维数为72.
5) 信号肽信息.我们通过SignalP [23] 程序从氨基酸序列中预测信号肽信息.对每个结构域,其得到的信号肽信息包含5 n 个元素,其中 n 为氨基酸序列长度.因为每个结构域的氨基酸序列长度往往不一样,我们通过多示例算法miFV [21] 将其转化为单个向量.对每个结构域,其信号肽信息SignalP特征维数为84.
6) 无序残基信息.我们通过DISOPRED程序 [24] 从氨基酸序列中预测无序区域信息.对每个结构域,其产生的无序残基信息包含2 n 个元素,其中 n 为氨基酸序列长度.因为每个结构域的氨基酸长度往往不一样,我们通过多示例算法miFV [21] 转化为单个向量.对每个结构域,其无序残基信息DISOPRED特征维数为84.
7) 物化属性.我们通过SciDBMaker程序 [25] 从氨基酸序列中预测物化属性信息,并通过标准的logistic函数将每个元素归一化到0~1之间.对每个结构域,其物化属性Pychem特征维数为59.
综上所述,对每个结构域,总的特征维数为743.
1.3.1 算法框架
本文使用的快速多示例多标记学习MIMLfast [14] 为一个2层的分类模型.在第1层中算法MIMLfast从GPCRs的原始特征空间学习出一个低维的子空间,该子空间是被所有GO标记共享;在第2层中,算法MIMLfast在基于GO标记共享的子空间为每个GO标记学习一个分类模型.这2层模型通过交替优化来拟合训练数据.
首先,对于GPCRs样本中的单一示例 x ,其在第 l 个GO标记上的分类模型为
![]()
(5)
其中, W 0 是一个 b × d 的共享子空间矩阵,目的是将原始GPCRs样本的特征维度由 d 维降到 b 维;而 w l 是对应于第 l 个GO标记的模型权重向量.这里 W 0 对应为模型的第1层,而 w l 对应为模型的第2层.
其次,如果GPCRs样本含有某个GO标记,是由于其中的某个示例含有该GO标记,即一个GPCRs的GO标记实际上由它的示例中最可能是正例的那个决定.因此,我们定义GPCRs样本在第 l 个GO标记上的预测值如下:
![]()
(6)
其中,示例 x 称为GPCRs样本 X 在第 l 个GO标记上的关键示例.对于GPCRs示例包 X 以及它的一个相关标记 y l , R ( X , l )的定义如下:
(7)
其中, ![]() 为GPCRs样本 X 的无关标记集合, I [·]是指示函数,当括号内为真时返回1,否则返回0; R ( X ; l )计算的是当前模型在包 X 上把多少个无关GO标记排在了相关GO标记 y l 的前面.
为GPCRs样本 X 的无关标记集合, I [·]是指示函数,当括号内为真时返回1,否则返回0; R ( X ; l )计算的是当前模型在包 X 上把多少个无关GO标记排在了相关GO标记 y l 的前面.
定义GPCRs样本 X 在GO标记 y l 上的排序错误如下:
![]() / i .
/ i .
(8)
由此可知,当GO标记 y l 被排的越靠后,排序错误 ε 越大.那么,整个GPCRs样本集上的排序错误为
(9)
其中, n 为样本个数, Y i 为第 i 个样本相关标记集合.我们的目标是要最小化式(9),使得所有的相关GO标记 y l 都尽量排在无关GO标记 ![]() 之前.根据式(7)可知,排序错误 ε ( X , l )实际上可以被分配到
之前.根据式(7)可知,排序错误 ε ( X , l )实际上可以被分配到 ![]() 中的每个无关GO标记上,如下:
中的每个无关GO标记上,如下:
![]()
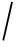 R ( X , l ),
R ( X , l ),
(10)
其中, I [·]是指示函数,当括号内为真时返回1,否则返回0.由于排序错误 ε ( X , l )是非凸非连续的,其优化问题是NP难问题,因此难于对式(10)进行直接优化.为解决此问题,我们采用hinge loss来代替:
![]()
f j ( X )- f l ( X )| + R ( X , l ),
(11)
其中,当 q ≥0时,| q | + = q ;否则,| q | + =0.
1.3.2 算法求解
算法训练过程中的每一轮,均从训练样本数据集中随机抽取一个多示例多标记GPCRs样本示例包 X 、 X 的一个相关GO标记 y 、一个无关GO标记 ![]() 然后根据所抽取的内容,使用随机梯度下降方法(SGD)优化算法的损失函数.
然后根据所抽取的内容,使用随机梯度下降方法(SGD)优化算法的损失函数.
假设在第 t 轮SGD中采样的三元组 ![]() 并且GO标记 y 上的关键示例为 x .相应地,无关GO标记
并且GO标记 y 上的关键示例为 x .相应地,无关GO标记 ![]() 的关键示例为
的关键示例为 ![]() 上述三元组上的损失函数为
上述三元组上的损失函数为

(12)
其中, ![]()
为了优化损失函数,须先求出函数 R ( X , y ),即针对每一个无关GO标记 ![]() 将
将 ![]() 与 f y ( X )进行对比,从而决定有多少无关GO标记排在了GO标记 y 前面.但是,当GO标记个数较大时,这个过程会非常耗时.本文,我们采取快速策略 [26] 得到 R ( X , y )的一个近似解.具体为,在每一轮随机梯度下降过程中,当确定了三元组中的 X 和 y 之后,我们一个一个地随机从无关GO标记子集
与 f y ( X )进行对比,从而决定有多少无关GO标记排在了GO标记 y 前面.但是,当GO标记个数较大时,这个过程会非常耗时.本文,我们采取快速策略 [26] 得到 R ( X , y )的一个近似解.具体为,在每一轮随机梯度下降过程中,当确定了三元组中的 X 和 y 之后,我们一个一个地随机从无关GO标记子集 ![]() 中抽样一个标记,直到出现了一个GO标记
中抽样一个标记,直到出现了一个GO标记 ![]() 排在了 y 前面为止,即满足
排在了 y 前面为止,即满足 ![]() 我们称这样排在相关GO标记之前的无关标记为违规标记.假设我们是在第 v 次抽样中找到了第1个违规标记,则 R ( X , y )可以被近似为
我们称这样排在相关GO标记之前的无关标记为违规标记.假设我们是在第 v 次抽样中找到了第1个违规标记,则 R ( X , y )可以被近似为 ![]() v .
v .
在第 t 轮SGD下降过程中,如果当前模型对无关GO标记的预测值小或等于相关GO标记的预测值,也即: ![]() 模型不更新;否则,根据式(13)进行更新模型:
模型不更新;否则,根据式(13)进行更新模型:
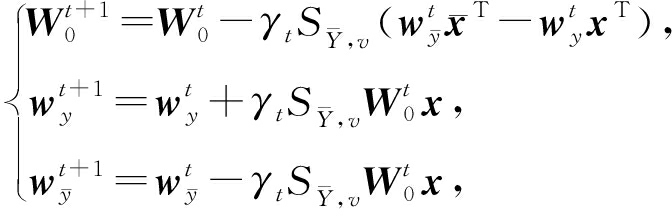
(13)
其中 γ t 为SGD算法更新的步长.
本文采用10倍交叉验证来评估模型,即把GPCRs样本随机分为10等分,将其中9等分用来训练模型,剩下1等分用来测试模型,重复10次,以保证每个GPCRs样本都被测试过1次.本文采用多示例多标记学习的评价指标,即Hamming loss( HL ),Ranking loss( RL ),OneError( OE )和Average precision( AP ) [5] . HL 用于考察多标记分类器分类错误的程度,评估样本在单个标记上的真实标记和预测标记之间的误差率; RL 用于考察在样本的预测标记排序序列中存在的排序错误,即样本的无关标记位于相关标记之前的次数; OE 用于考察在样本的真实标记的排序序列中,序列最前端即排在第1的标记不在样本的预测标记集合中的次数; AP 用来考察在样本的预测标记排序序列中,位于样本的某个相关标记之前的标记仍为相关标记的比例,反映了预测标记的平均精确度, AP 越大,表示模型性能越好,其他评价指标相反.
表1显示了GPCRs结构域各种特征对模型预测性能的贡献.由表1可以看出,对GO分子功能, GPCRs结构域特征对模型 AP 的贡献由大到小依次为Conjoint Triad>PSSMs>AAC>DISOPRED>SSC>Pychem>SignalP;而对GO生物学过程,其特征的贡献由大到小依次为Conjoint triad>AAC>PSSMs>SSC>DISOPRED>Pychem>SignalP.当使用所有的特征时(对应于ALL列),我们的模型得到了最好的性能.例如,对GO分子功能,模型的 AP =0.886 8;对GO生物学过程,模型的 AP =0.881 8.
Table 1 Contributions of Domain Features on Predictive Performance
表1 结构域特征对模型预测性能的贡献

Notes: “↑” indicates the larger the value, the better the performance; “↓” indicates the smaller the value, the better the performance; the best results on each evaluation criterion are highlighted in boldface.
我们与4种多示例多标记学习方法进行了比较,分别是MIMLRBF [27] ,ENMIMLNNBP [3] ,MIMLKNN [5] ,MIMLSVM [4] .MIMLfast和4种对比方法均采用参考文献中的默认参数.对MIMLfast方法,共享空间维度设为100;对MIMLRBF方法,缩放因子设为0.08;对MIMLKNN方法,簇的数量设为样本数量的40%;对MIMLSVM方法,高斯核半径 r =0.2; 对ENMIMLNNBP方法,学习率设为0.4.表2显示了我们的模型与多示例多标记学习方法的比较结果(基于所有的特征).其中↑表示评价指标值越大,性能越好,↓则相反;最优的结果用粗体标注.从表2可知,在GPCRs的GO分子功能和生物学过程预测上,我们的方法均优于4种基于多示例多标记学习的预测方法.例如,对GO分子功能,我们模型比次优的ENMIMLNNBP方法高0.071 9( AP 值);对GO生物学过程,我们模型比次优的MIMLRBF方法高0.136 7( AP 值).
效率是多示例多标记学习的一个瓶颈,因此对方法的效率进行检验非常关键.本文的实验全部在一台具有4×2.60 GHz CPU 和16 GB内存的PC机上完成.表3给出了各个多示例多标记学习算法在分子功能和生物学过程数据集上的时间开销比较.从表3可以看出,我们的方法在2个数据集上都是效率最高的,远远少于比较方法的时间开销.Huang等人(2014)论文中的多个结果显示,本文基于的MIMLfast方法的时间复杂性与样本数量呈现一个线性相关,在处理这么大规模数据具有很大的优势 [14] .
Table 2 Comparison with Multi - Instance Multi - label Learning Based Methods
表2 与多示例多标记学习方法的比较

Notes: “↑” indicates the larger the value, the better the performance; “↓” indicates the smaller the value, the better the performance; the best results on each evaluation criterion are highlighted in boldface.
Table 3 Runtime Comparison with Multi - Instance Multi - label Learning Based Methods
表3 与多示例多标记学习方法的时间效率比较 s

本文与2种多标记学习方法进行比较,分别是BPMLL [28] 和ML-KNN [29] ,它们均采用参考文献中的默认参数.即对BPMLL方法,学习率设为0.05;对ML-KNN方法,近邻样本个数设为10.我们将多示例转化为单示例采用了3种方法:miFV [21] ,miVLAD [21] 和平均(Means).平均的方法就是将每个样本包的所有示例求平均,得到单示例向量.表4显示了我们的模型与多标记学习算法的比较结果.其中↑代表评价指标越大,性能越好,↓则相反;最优的结果用粗体进行标注.表4显示我们的算法优于所有的多标记学习方法,例如,我们模型比最优的多标记学习BPMLL(基于miFV单示例化方法)在GO分子功能上高0.288 1( AP 值)和在GO生物学过程上高0.310 9( AP 值).这或许表明将多示例转换为单示例过程中会损失较多信息.
Table 4 Comparison with Multi - Label Learning Based Methods
表4 与多标记学习方法的比较

Notes: “↑” indicates the larger the value, the better the performance; “↓” indicates the smaller the value, the better the performance; the best results on each evaluation criterion are highlighted in boldface.
Critical Assessment of Functional Annotation(CAFA)是国际上最权威针对蛋白质功能进行注释的比赛,到现在已经举行了3届.本文中,我们与多个CAFA比赛中蛋白质功能预测方法进行比较,包括FFPred [30] ,Argot [31] ,INGA [32] ,我们通过这3个方法的在线预测平台得到GPCRs的预测结果.为了对比的公平性,我们把平台得到的预测结果,根据2.1节中的go.obo文件补充其父节点GO术语,补充之后的GO术语与训练样本中的GO术语标记空间一样.
表5显示了与CAFA蛋白质功能预测方法的比较结果.其中↑代表评价指标越大,性能越好,↓则相反;最优的结果用粗体进行标注.我们采用了不同的阈值( cuto f f )来区分正负样本,即预测值大于等于 cuto f f ,为正样本,否则为负样本.实验结果表明,在GPCRs的GO分子功能和生物学过程预测上,我们的方法均优于这3种CAFA蛋白质功能预测方法,如表5所示.例如,我们模型比最优的CAFA蛋白质功能预测方法Argot在GO分子功能上高0.265 3( AP 值)和在GO生物学过程上高0.291 3( AP 值).
Table 5 Comparison with CAFA Protein Function Prediction Methods
表5 与CAFA蛋白质功能预测方法的比较
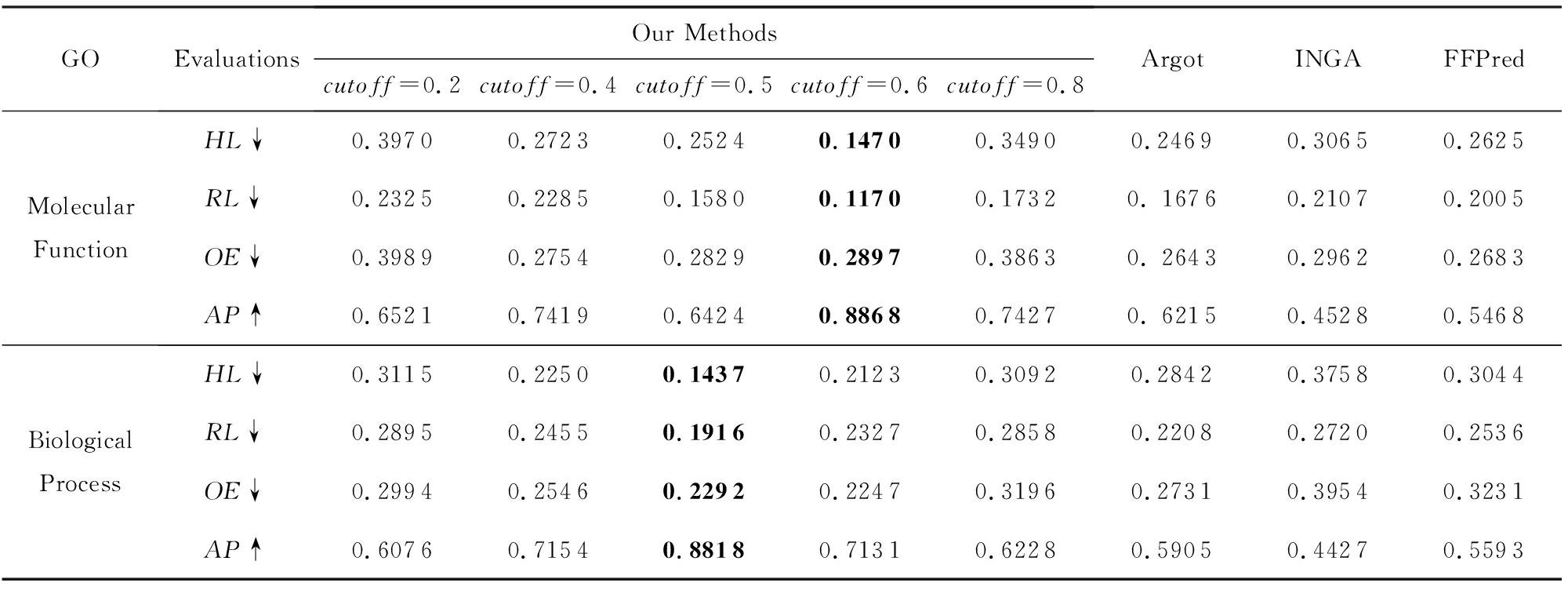
Notes: “↑” indicates the larger the value, the better the performance; “↓” indicates the smaller the value, the better the performance; the best results on each evaluation criterion are highlighted in boldface.
本文提出了一种基于快速多示例多标记学习MIMLfast的G蛋白偶联受体的生物学功能预测方法,该方法采用了一种新的混合特征,它考虑了GPCRs结构域的三联氨基酸、氨基酸关联、进化、二级结构关联、信号肽、无序残基等多种信息.方法包括2个步骤:1)基于G蛋白偶联受体的特征空间,学习出一个低维的子空间,该子空间为所有GO标记所共享;2)为每个GO标记根据子空间中相关标记信息,学习出一个分类模型.实验结果证明,我们的模型获得了很好的性能,优于目前最优的多示例多标记学习、多标记学习的预测方法和CAFA蛋白质功能预测方法.
参考文献
[1]Miller W, Lefkowitz R. Expanding roles for β -arrestins as scaffolds and adapters in GPCR signaling and trafficking[J]. Current Opinion in Cell Biology, 2001, 13(2): 139-145
[2]Overington J, Al-Lazikani B, Hopkins A. How many drug targets are there?[J]. Nature Reviews Drug Discovery, 2007, 5(12): 993-996
[3]Wu Jiansheng, Huang Shengjun, Zhou Zhihua. Genome-wide protein function prediction through multi-instance multi-label learning[J]. IEEE  ACM Trans on Computational Biology & Bioinformatics, 2014, 11(5): 891-902
ACM Trans on Computational Biology & Bioinformatics, 2014, 11(5): 891-902
[4]Zhou Zhihua, Zhang Minling. Multi-instance multi-label learning with application to scene classification[C] Proc of Int Conf on Neural Information Processing Systems (NIPS’06). Cambridge, MA: MIT Press, 2006: 1609-1616
[5]Zhou Zhihua, Zhang Minling, Huang Shengjun, et al. Multi-instance multi-label learning[J]. Artificial Intelligence, 2012, 176(1): 2291-2320
[6]Nguyen N. A new SVM approach to multi-instance multi-label learning[C] Proc of IEEE Int Conf on Data Mining (ICDM’11). Piscataway, NJ: IEEE, 2011: 384-392
[7]Zhang Minling, Zhou Zhihua. M3MIML: A maximum margin method for multi-instance multi-label learning[C] Proc of IEEE Int Conf on Data Mining (ICDM’08). Piscataway, NJ: IEEE, 2008: 688-697
[8]Zhang Minling, Zhou Zhihua.Multi-label learning by instance differentiation[C] Proc of AAAI Conf on Artificial Intelligence (AAAI’07). Palo Alto, CA: AAAI, 2007: 669-674
[9]Yang Shuanghong, Zha Hongyuan, Hu Baogang. Dirichlet-bernoulli alignment: A generative model for multi-class multi-label multi-instance corpora[C] Proc of Advances in Neural Information Processing Systems (NIPS’09). Cambridge, MA: MIT Press, 2009: 2143-2150
[10]Jin Rong, Wang Shijun, Zhou Zhihua. Learning a distance metric from multi-instance multi-label data[C] Proc of Computer Vision and Pattern Recognition (CVPR’09). Piscataway, NJ: IEEE, 2009: 896-902
[11]Briggs F, Fern X, Raich R. Rank-loss support instance machines for MIML instance annotation[C] Proc of ACM SIGKDD Int Conf on Knowledge Discovery and Data Mining (KDD’09). New York: ACM, 2009: 534-542
[12]Radivojac P, Clark W, Oron T, et al. A large-scale evaluation of computational protein function prediction[J]. Nature Methods, 2013, 10(3): 221-229
[13]Ashburner M, Ball C A, Blake J, et al. Gene ontology: Tool for the unification of biology[J]. Nature Genetics, 2000, 25(1): 25-29
[14]Huang Shengjun, Gao Wei, Zhou Zhihua. Fast multi-instance multi-label learning[C] Proc of AAAI Conf on Artificial Intelligence (AAAI’14). Palo Alto, CA: AAAI, 2014: 1868-1874
[15]Bairoch A, Apweiler R, Wu C, et al. The universal protein resource (UniProt)[J]. Nucleic Acids Research, 2008, 36(Database issue): D190-D195
[16]Camacho C, Coulouris G, Avagyan V, et al. BLAST+: Architecture and applications[J]. BMC Bioinformatics, 2009, 10(1): 421-429
[17]Marchlerbauer A, Anderson J, Chitsaz F, et al. CDD: Specific functional annotation with the Conserved Domain Database[J]. Nucleic Acids Research, 2009, 37(1): D205-D210
[18]Wu Jiansheng, Liu Hongde, Duan Xueye, et al. Prediction of DNA-binding residues in proteins from amino acid sequences using a random forest model with a hybrid feature[J]. Bioinformatics, 2009, 25(1): 30-35
[19]Wu Jiansheng, Hu Dong, Xu Xin, et al. A novel method for quantitatively predicting non-covalent interactions from protein and nucleic acid sequence[J]. Journal of Molecular Graphics & Modelling, 2011, 31(11): 28-34
[20]Liu Zhihua, Meng Jihong, Sun Xiao. A novel feature-based method for whole genome phylogenetic analysis without alignment: Application to HEV genotyping and subtyping[J]. Biochemical & Biophysical Research Communications, 2008, 368(2): 223-30
[21]Wei Xiushen, Wu Jianxin, Zhou Zhihua. Scalable algorithms for multi-instance learning[J]. IEEE Trans Neural Networks Learning Systems, 2017, 28(4): 975-987
[22]Mcguffin L, Bryson K, Jones D. The PSIPRED protein structure prediction server[J]. Bioinformatics, 2000, 16(4): 404-405
[23]Petersen T, Brunak S, Von H, et al. SignalP 4.0: Discriminating signal peptides from transmembrane regions[J]. Nature Methods, 2011, 8(10): 785-786
[24]Jones D, Cozzetto D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity[J]. Bioinformatics, 2015, 31(6): 857-863
[25]Hammami R, Zouhir A, Naghmouchi K, et al. SciDBMaker: New software for computer-aided design of specialized biological databases[J]. BMC Bioinformatics, 2008, 9(1): 121-126
[26]Weston J, Bengio S, Usunier N. WSABIE: Scaling up to large vocabulary image annotation[C] Proc of Int Joint Conf on Artificial Intelligence (IJCAI’11). San Mateo, CA: Morgan Kaufmann, 2011: 2764-2770
[27]Zhang Minling, Wang Zhijian. MIMLRBF: RBF neural networks for multi-instance multi-label learning[J]. Neurocomputing, 2009, 72(16): 3951-3956
[28]Zhang Minling, Zhou Zhihua. Multilabel neural networks with applications to functional genomics and text categorization[J]. IEEE Trans on Knowledge and Data Engineering, 2006, 18(10): 1338-1351
[29]Zhang Minling, Zhou Zhihua. ML-KNN: A lazy learning approach to multi-label learning[J]. Pattern Recognition, 2007, 40(7): 2038-2048
[30]Domenico C, Federico M, Hannah C, et al. FFPred 3: Feature-based function prediction for all gene ontology domains[J]. Scientific Reports, 2016, 6(8): 31865-31875
[31]Falda M, Toppo S, Pescarolo A, et al. Argot2: A large scale function prediction tool relying on semantic similarity of weighted gene ontology terms[J]. BMC Bioinformatics, 2012, 13(Suppl): S14
[32]Piovesan D, Giollo M, Leonardi E, et al. INGA: Protein function prediction combining interaction networks, domain assignments and sequence similarity[J]. Nucleic Acids Research, 2015, 43(1): 134-140

Wu Jiansheng , born in 1979. PhD, associate professor. His main research interests include machine learning and bioinformatics.

Feng Qiaoyu , born in 1991. Master. Her main research interest is machine learning.

Yuan Jingzhou , born in 1997. Undergraduate in biomedical engineering. His main research interest is biomedical hardware.

Hu Haifeng , born in 1973. PhD, associate professor. His main research interests include large-scale similarity search, wireless sensor networks, wireless networking and distributed systems.

Zhou Jiate , born in 1997. Undergraduate in biomedical engineering. His main research interests include biomedical imaging and image processing.

Gao Hao , born in 1997. Undergraduate in biomedical engineering. His main research interests include biomedical hardware and software developing.
Wu Jiansheng 1 , Feng Qiaoyu 2 , Yuan Jingzhou 1 , Hu Haifeng 2 , Zhou Jiate 1 , and Gao Hao 1
1 ( School of Geographic and Biological Information , Nanjing University of Posts and Telecommunications , Nanjing 210023) 2 ( School of Telecommunication and Information Engineering , Nanjing University of Posts and Telecommunications , Nanjing 210003)
Abstract G protein-coupled receptors (GPCRs) constitute the largest family among human membrane proteins which are the important targets of many drugs on the market. An accurate annotation of the biological functions of GPCR proteins is key to understand their involved biological processes and drug-acting mechanisms. In our previous work, we found that protein function prediction problem can be formulated as a multi-instance multi-label learning (MIML) task. In this paper, we propose a novel method for predicting biological functions of G protein-coupled receptors by using a fast MIML learning called MIMLfast along with a hybrid feature. The hybrid feature consists of amino acid triple information, amino acid correlation information, evolutionary information, secondary structure correlation information, signal peptide information, disordered residue information, physical and chemical properties among GPCR domains. The experimental results show that our method achieves good performance which is superior to state-of-the-art multi-instance multi-label learning methods, multi-label learning methods and CAFA protein function prediction methods.
Key words G protein-coupled receptors (GPCRs); predicting biological functions; fast multi-instance multi-label learning; domains; hybrid feature
中图法分类号 TP315.69
This work was supported by the National Natural Science Foundation of China (81771478, 61571233), the Key University Science Research Project of Jiangsu Province (17KJA510003), and the Natural Science Foundation of Nanjing University of Posts and Telecommunications (NY218092).
基金项目 : 国家自然科学基金项目(81771478,61571233);江苏省高等学校自然科学研究项目(17KJA510003);南京邮电大学科研基金项目(NY218092)
收稿日期 : 2018-05-18;
修回日期: 2018-06-07
DOI: 10.7544/issn1000-1239.2018.20180361